Clear Sky Science · it
Un benchmark conformazionale per la predizione delle proprietà ottiche con reti neurali su grafi sensibili al solvente
Perché è importante predire i colori delle molecole
Dai pixel vivaci negli schermi dei telefoni ai coloranti nelle celle solari e alle sonde fluorescenti usate per osservare i tessuti viventi, molte tecnologie moderne dipendono da molecole che assorbono ed emettono luce a colori ben precisi. Progettare queste molecole è difficile: piccole modifiche nella struttura o nel solvente possono spostare drasticamente i loro colori, e i calcoli tradizionali di chimica quantistica sono troppo lenti per guidare ricerche su larga scala. Questo articolo presenta un nuovo dataset e modelli di machine learning che trattano le molecole in pieno tre dimensioni e tengono esplicitamente conto dell’ambiente liquido circostante, permettendo predizioni delle proprietà ottiche molto più rapide e accurate.
Costruire una mappa migliore delle molecole colorate
Gli autori hanno innanzitutto raccolto e ripulito un’ampia collezione di dati sperimentali su come i «cromofori» organici — le parti delle molecole che assorbono la luce — si comportano in diversi solventi. Hanno combinato diversi dataset pubblici e poi corretto con cura problemi come descrizioni strutturali invalide, stati di carica incoerenti e strutture contenenti metalli fuorvianti. Il risultato è nablaColors, che copre 13.731 molecole uniche e 26.369 coppie cromoforo–solvente con misure di assorbimento e, per molte di esse, anche lunghezze d’onda di emissione e efficienza di emissione luminosa (resa quantica di fotoluminescenza). Questa curatela accurata riduce il rumore che può confondere i modelli di machine learning e stabilisce una base affidabile per studi successivi.
Aggiungere la terza dimensione mancante
La maggior parte degli strumenti di machine learning esistenti per predire proprietà ottiche rappresentano le molecole come grafi bidimensionali: gli atomi sono nodi e i legami chimici sono linee. Tuttavia, gli stati eccitati e l’assorbimento della luce dipendono sensibilmente dalle vere forme tridimensionali — angoli di legame, torsioni e interazioni deboli — che queste rappresentazioni 2D non riescono a catturare appieno. Per rimediare, il gruppo ha generato strutture 3D per ogni cromoforo usando una pipeline a più fasi: un primo layout 3D approssimativo, un metodo semi-empirico quantistico più veloce e poi ottimizzazioni più accurate con teoria del funzionale della densità (DFT), sia in vuoto sia con un modello implicito del solvente circostante. Questa nuova estensione 3D, nablaColors-3D, fornisce più conformazioni per molecola, ciascuna riflettendo un diverso livello di realismo fisico e costo computazionale. 
Insegnare alle reti neurali a vedere forma e solvente
Con nablaColors-3D a disposizione, gli autori hanno costruito un benchmark per confrontare una gamma di modelli di machine learning, dalle affermate reti neurali su grafi 2D alle architetture 3D all’avanguardia che rispettano le simmetrie fisiche nello spazio. Hanno inoltre progettato un aggiornamento «sensibile al solvente»: una rete neurale separata e leggera codifica la struttura del solvente a partire dalla sua rappresentazione molecolare, producendo un’impronta compatta del solvente. Questa impronta viene combinata con la rappresentazione 3D del cromoforo in modo che il modello principale possa apprendere come l’ambiente liquido modifichi sottilmente la geometria e la struttura elettronica della molecola. Utilizzando una rigorosa divisione dei dati basata sullo scaffold, il benchmark assicura che molecole strettamente correlate non compaiano né nel set di addestramento né in quello di test, così le prestazioni misurate riflettono una vera generalizzazione piuttosto che semplice memorizzazione.
Quanti dettagli geometrici sono necessari?
Una domanda pratica importante è se valga la pena pagare l’alto costo computazionale di geometrie molto accurate. Il gruppo ha variato sistematicamente il tipo di conformazioni 3D fornite a ciascun modello — che vanno da strutture semi-empiriche più economiche a ottimizzazioni DFT più onerose in vuoto e in solvente implicito — mantenendo fissi tutti i parametri di addestramento. In generale, geometrie migliori tendevano a migliorare le predizioni, ma l’effetto dipendeva dal modello e dall’uso o meno di impronte esplicite del solvente. Una volta incluse le embedding del solvente, le differenze di prestazione tra le fonti geometriche si sono ridotte, mostrando che gran parte dell’influenza del solvente può essere catturata tramite questa codifica separata piuttosto che con calcoli sempre più costosi dei conformeri. Per il loro miglior modello hanno persino dimostrato che strutture poco costose generate da software chimici standard possono sostituire quelle ottimizzate quantisticamente durante l’addestramento con quasi nessuna perdita di accuratezza. 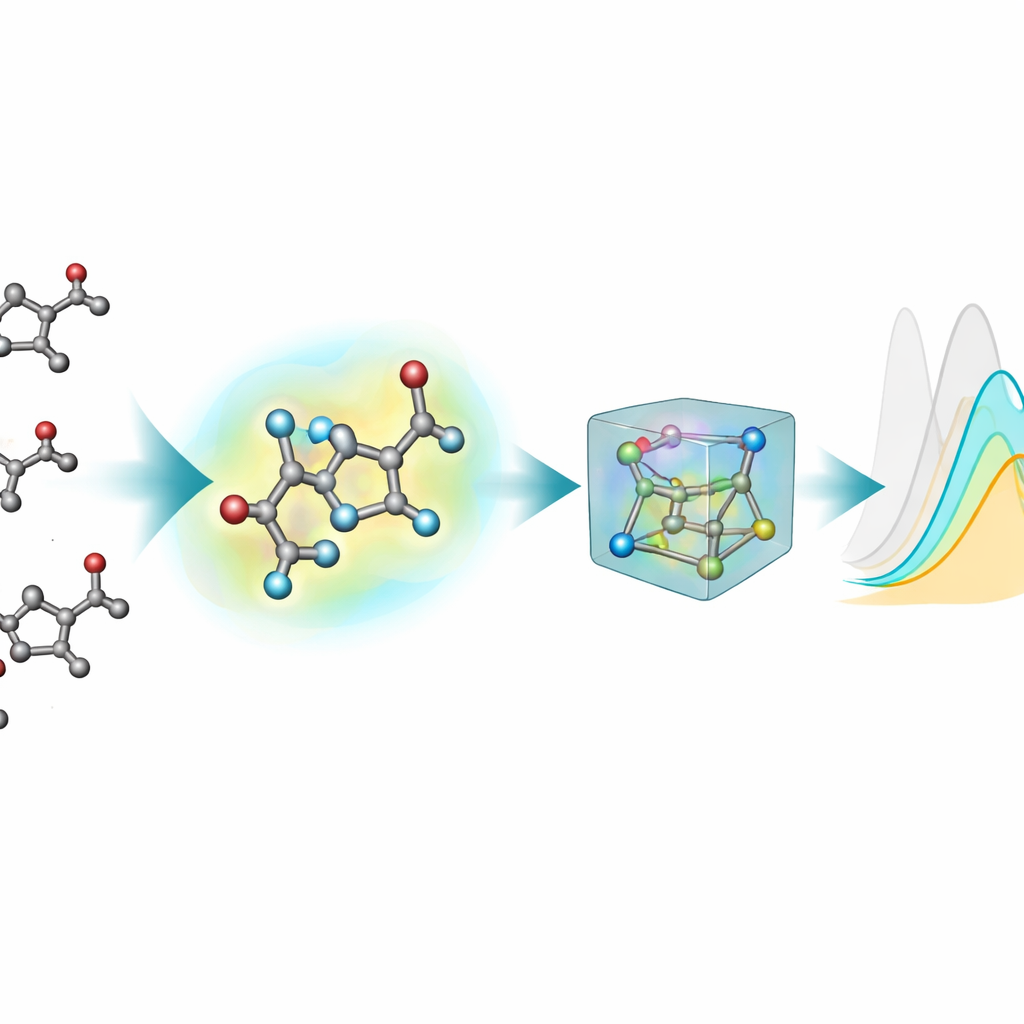
Un salto oltre i metodi tradizionali
Tra tutti i modelli testati, un’architettura 3D basata su transformer chiamata UniMol+ — aumentata con embedding del solvente in una variante che gli autori chiamano UniProp — ha ottenuto le migliori prestazioni. UniProp ha raggiunto un errore assoluto medio di circa 16 nanometri per le lunghezze d’onda di assorbimento su un set di test non utilizzato durante l’addestramento, oltre il 30% in meno rispetto al miglior baseline 2D e di gran lunga migliore rispetto a un metodo DFT dipendente dal tempo largamente usato, che sbagliava di circa 62 nanometri. Crucialmente, UniMol+ era stato pre-addestrato su grandi dataset di chimica quantistica per imparare a raffinare strutture 3D approssimative verso geometrie di livello elevato. Questa capacità di «denoising geometrico» le permette di accettare conformeri relativamente economici al momento della predizione pur catturando i dettagli strutturali fini che contano per il comportamento ottico.
Verso uno strumento universale per il design ottico
Infine, gli autori hanno esteso UniProp per predire non solo i picchi di assorbimento, ma anche le lunghezze d’onda di emissione e l’efficienza di emissione luminosa in un unico modello multitarget. Ha mantenuto un’elevata accuratezza su tutte e tre le proprietà, con solo un lieve compromesso per l’assorbimento, dimostrando che le stesse caratteristiche 3D catturano fattori fisici comuni dietro diversi processi fotofisici. Per i non specialisti, il punto chiave è che reti neurali tridimensionali e sensibili al solvente — addestrate su un benchmark curato con attenzione — possono ora superare i metodi quantistici tradizionali offrendo, allo stesso tempo, velocità di esecuzione ordini di grandezza superiori. Questo rende realistico lo screening virtuale di enormi librerie di coloranti candidati, emettitori OLED e sonde fluorescenti, accelerando la scoperta di molecole con colori e luminosità finemente tarati.
Citazione: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Parole chiave: ottica molecolare, reti neurali su grafi, machine learning per la chimica, coloranti fluorescenti, effetti del solvente