Clear Sky Science · sv
Tolkningsbar, flexibel och rumsligt medveten integrering av flera spatiala transkriptomik-dataset från olika källor
Att se vävnader som levande kartor
Biologer har idag verktyg som läser vilka gener som är aktiva i tusentals små punkter över ett vävnadssnitt och förvandlar organ, tumörer och embryon till detaljerade molekylära kartor. Men varje experiment använder olika instrument och inställningar, vilket gör att de resulterande kartorna är svåra att jämföra eller slå ihop. Denna artikel presenterar INSPIRE, en beräkningsmetod som sammanfogar dessa olika genkartor till en enda begriplig vy och hjälper forskare att följa hur vävnader byggs upp, förändras vid sjukdom och utvecklas över tid.
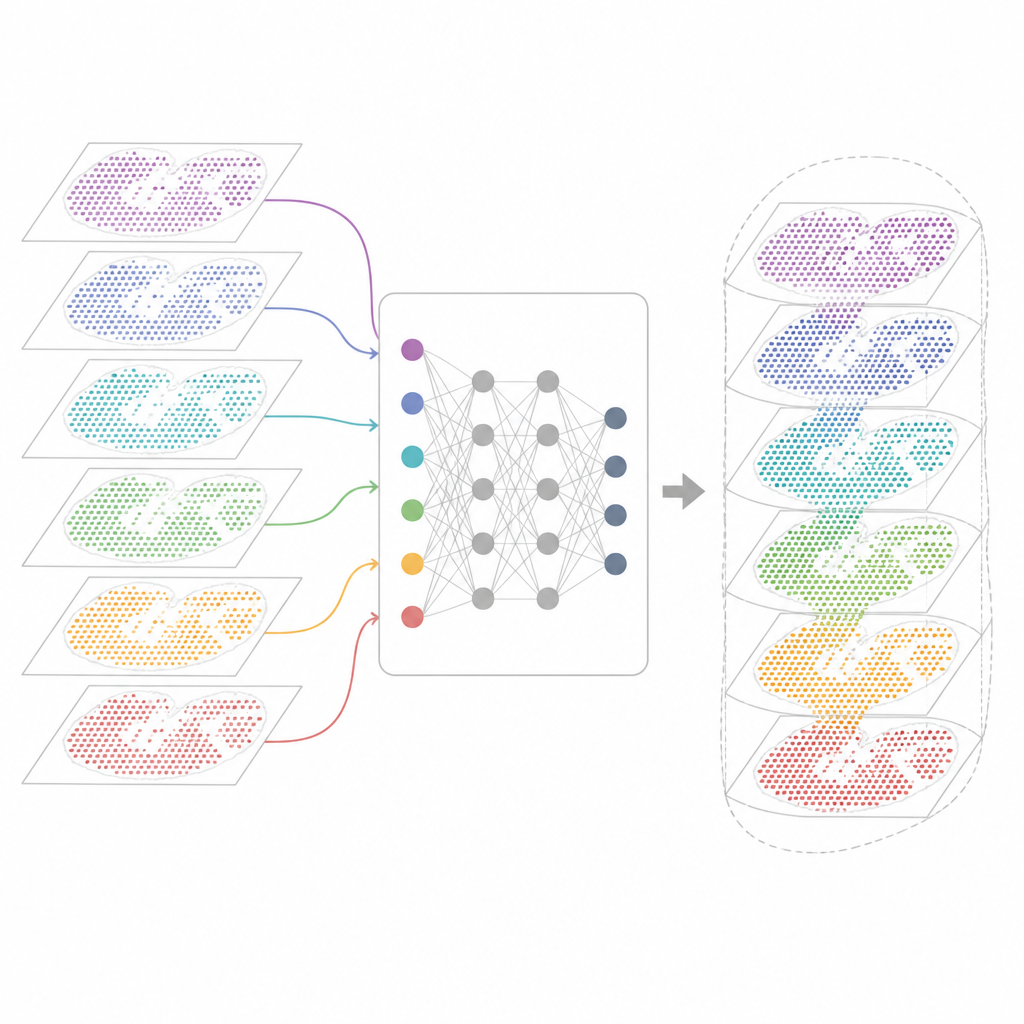
Varför många genkartor är svåra att kombinera
Moderna spatiala transkriptomiktekniker mäter genaktivitet samtidigt som de bevarar var varje cell ligger i vävnaden. Vissa metoder fångar nästan alla gener men suddar ut flera celler i varje punkt. Andra prickar in enstaka celler men bara för en utvald panel av gener. Dessa kartor kommer också från olika laboratorier, maskiner, tidpunkter och arter. Följaktligen bär varje dataset sina egna tekniska egendomligheter och brus. Befintliga analysverktyg kan beskriva ett snitt eller några få liknande snitt, men de misslyckas ofta när de ska justera flera dussin sektioner, överbrygga olika teknologier eller hålla isär både delade och unika vävnadsdrag.
Ett nytt ramverk för att förena vävnadskartor
INSPIRE tar itu med detta integrationsproblem med ett djupinlärningsramverk som respekterar både genavläsningarna och cellernas fysiska läge. Först bygger det en "nabohetsgraf" för varje vävnadssnitt som kopplar samman närliggande punkter. Ett grafbaserat neuralt nätverk omvandlar sedan rådata till en gemensam intern representation som blandar jämförbara celler från olika sektioner samtidigt som sektionsspecifika mönster får finnas kvar. En adversariell komponent fungerar som en kritiker som upptäcker var sektioner ännu inte överlappar väl i detta interna utrymme och skjuter modellen mot bättre anpassning.
Från dolda mönster till tolkningsbara egenskaper
När data väl är förda in i detta delade utrymme bryter INSPIRE ned den kvarvarande signalen i en uppsättning återkommande spatiala mönster, kallade faktorer, var och en kopplad till ett karakteristiskt genprogram. Detta görs med ett steg av icke-negativ matrixfaktorisering som uppmuntrar modellen att representera data som en kombination av ett fåtal enkla byggstenar. Varje faktor motsvarar ett spatialt mönster över snitten, såsom ett specifikt hjärnlager, en tumörnisch eller ett utvecklande organs region. Eftersom INSPIRE också lär sig vilka gener som är mest bundna till varje faktor kan forskare tolka dessa mönster i termer av kända celltyper och biologiska processer, istället för abstrakta tal.
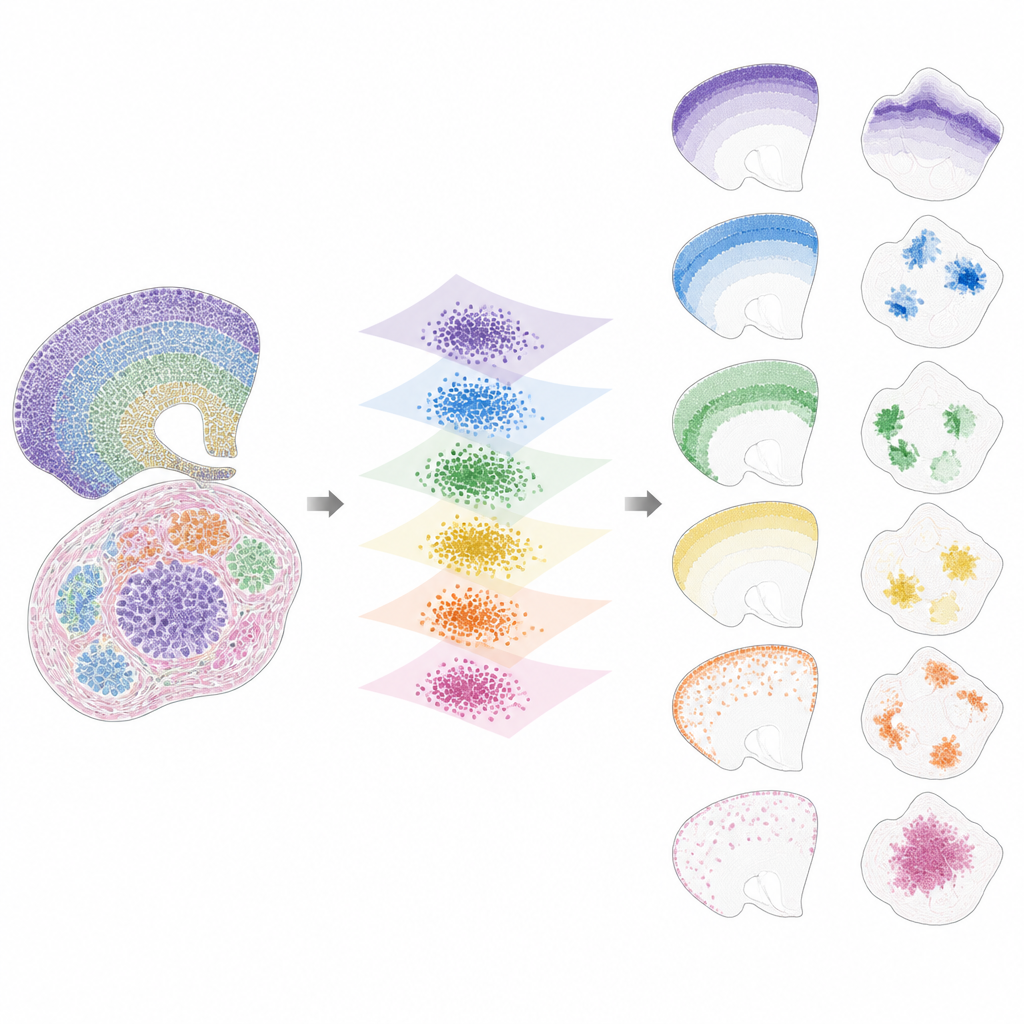
Vad INSPIRE avslöjar i verkliga dataset
Författarna visar att INSPIRE överträffar en rad populära metoder på både simulerade och verkliga dataset. I människans hjärnbarkssnitt justerar det lager över donatorer och återfinner fina skillnader mellan neurontyper och stödjeceller, inklusive subtila arrangemang som manuella etiketter missade. I musens hjärna separerar det tydligt delade strukturer, såsom cortex, från unika regioner som cerebellum, samtidigt som det korrekt identifierar deras distinkta genprofiler. INSPIRE överbrygger också olika teknologier genom att kombinera encellskartor med bredare undersökningar för att överföra detaljerad lagrinformation och för att härleda saknade genmönster som aldrig mättes direkt.
Följa sjukdom, utveckling och 3D-struktur
Utöver friska vävnader exponerar INSPIRE dold variation i sjukdom och utveckling. I human bröstcancer med sektioner som innehåller hundratusentals celler skiljer den icke-invasiva från invasiva tumörområden åt och upptäcker distinkta undergrupper av stödjeceller runt vardera, kopplade till kända markörer för aggressivitet och blodkärlstillväxt. I musembryon integrerar INSPIRE snitt tagna vid flera stadier för att bygga en spatiotemporal atlas som följer hur organ som hjärta, lever, lunga och hjärna växer och omorganiseras. Genom att noggrant justera intilliggande snitt stödjer den också tredimensionella rekonstruktioner av organ och hela embryon och förvandlar staplar av tvådimensionella bilder till sammanhängande 3D‑modeller av genuttryck.
Vad detta betyder för framtida vävnadsstudier
För en icke-specialist kan INSPIRE ses som en kraftfull översättare som förvandlar många ofullkomliga, brusiga genkartor till ett gemensamt språk av spatiala mönster och genprogram. Den bevarar kontexten för var celler ligger i vävnaden, filtrerar bort tekniska artefakter och lyfter fram både gemensamma och unika drag över experiment. När spatiala transkriptomikprojekt skalar upp till hela organ, tumörer och organismer kommer metoder som INSPIRE att vara nyckeln till att bygga integrerade atlaser som forskare kan utforska för att förstå hur celler samverkar, hur sjukdomar rubbar vävnadsarkitekturen och hur komplexa strukturer uppstår under utvecklingen.
Citering: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Nyckelord: spatial transkriptomik, vävnadsarkitektur, genuttryckskartor, dataintegration, tumörmikromiljö