Clear Sky Science · it
Integrazione interpretabile, flessibile e spazialmente consapevole di più dataset di trascrittomica spaziale provenienti da fonti diverse
Vedere i tessuti come mappe viventi
I biologi dispongono oggi di strumenti che leggono quali geni sono attivi in migliaia di minuscoli punti di una sezione tissutale, trasformando organi, tumori ed embrioni in mappe molecolari dettagliate. Tuttavia ogni esperimento usa strumenti e configurazioni diverse, quindi le mappe risultanti sono difficili da confrontare o combinare. Questo articolo presenta INSPIRE, un metodo computazionale che cuce insieme queste mappe geniche eterogenee in una visione singola e comprensibile, aiutando gli scienziati a seguire come i tessuti si costruiscono, cambiano con la malattia e si evolvono nel tempo.
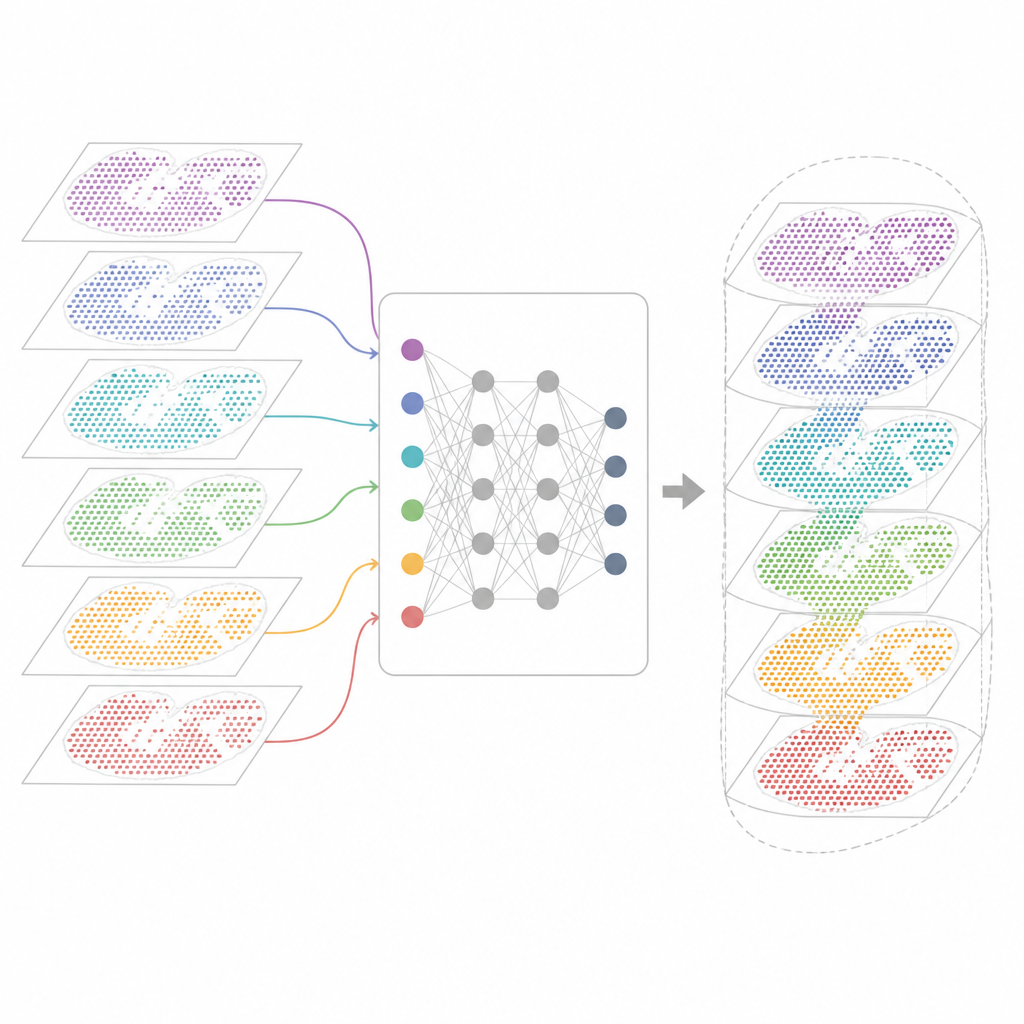
Perché molte mappe geniche sono difficili da combinare
Le tecnologie moderne di trascrittomica spaziale misurano l'attività genica preservando la posizione di ogni cellula nel tessuto. Alcuni metodi catturano quasi tutti i geni ma sovrappongono più cellule in ciascun punto. Altri individuano singole cellule ma solo per un pannello selezionato di geni. Queste mappe provengono anche da laboratori, macchine, tempi e specie diversi. Di conseguenza ciascun dataset porta con sé peculiarità tecniche e rumore propri. Gli strumenti di analisi esistenti possono descrivere una singola sezione o poche sezioni simili, ma spesso falliscono quando devono allineare decine di sezioni, collegare tecnologie diverse o tenere traccia contemporaneamente di caratteristiche tissutali condivise e specifiche.
Un nuovo quadro per unificare le mappe tissutali
INSPIRE affronta questo problema di integrazione con un framework di deep learning che rispetta sia i dati di espressione genica sia la disposizione fisica delle cellule. Costruisce innanzitutto un "grafo di vicinato" per ciascuna sezione tissutale, connettendo i punti vicini. Una rete neurale basata su grafi poi trasforma i dati grezzi in una rappresentazione interna condivisa che mescola cellule comparabili provenienti da sezioni diverse pur permettendo che rimangano pattern specifici delle singole sezioni. Un componente avversario agisce come un critico, individuando dove le sezioni non si sovrappongono ancora bene in questo spazio interno e spingendo il modello a migliorare l'allineamento.
Da pattern nascosti a caratteristiche interpretabili
Una volta che i dati sono stati portati in questo spazio condiviso, INSPIRE scompone il segnale residuo in un insieme di schemi spaziali ricorrenti, chiamati fattori, ciascuno collegato a un programma genico caratteristico. Questo avviene mediante un passo di fattorizzazione in matrici non negative che incoraggia il modello a rappresentare i dati come combinazione di pochi blocchi di costruzione semplici. Ogni fattore corrisponde a uno schema spaziale attraverso le sezioni, come un particolare strato cerebrale, una nicchia tumorale o una regione di un organo in sviluppo. Poiché INSPIRE impara anche quali geni sono più associati a ciascun fattore, i ricercatori possono interpretare questi schemi in termini di tipi cellulari e processi biologici noti, anziché numeri astratti.
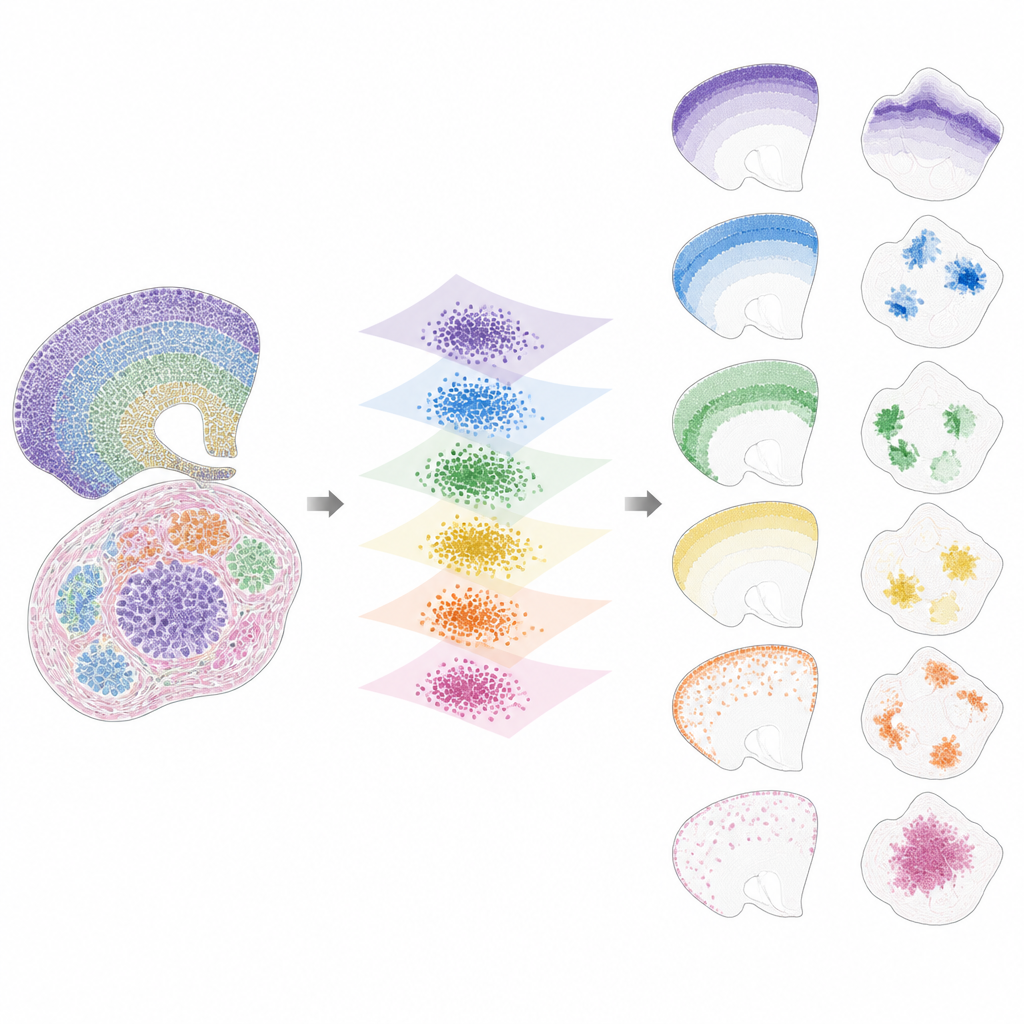
Cosa rivela INSPIRE in dataset reali
Gli autori mostrano che INSPIRE supera una serie di metodi popolari sia su dataset simulati sia reali. In sezioni della corteccia cerebrale umana, allinea gli strati tra i donatori e recupera sottili distinzioni tra tipi neuronali e cellule di supporto, incluse disposizioni delicate che le etichette manuali avevano mancato. Nel cervello di topo separa nettamente strutture condivise, come la corteccia, da regioni uniche come il cervelletto, identificandone correttamente le distintive firme geniche. INSPIRE inoltre mette in collegamento tecnologie diverse, combinando mappe a singola cellula con indagini più ampie per trasferire informazioni dettagliate sugli strati e per inferire pattern genici mancanti che non erano stati direttamente misurati.
Seguire malattia, sviluppo e struttura 3D
Oltre ai tessuti sani, INSPIRE mette in luce variazioni nascoste nella malattia e nello sviluppo. In sezioni di carcinoma mammario umano con centinaia di migliaia di cellule, distingue regioni tumorali non invasive da quelle invasive e scopre sottogruppi distinti di cellule di supporto attorno a ciascuna, collegati a marcatori noti di aggressività e crescita vascolare. Negli embrioni di topo, INSPIRE integra sezioni prese a stadi multipli per costruire un atlante spazio-temporale, tracciando come organi come cuore, fegato, polmone e cervello crescono e si riorganizzano. Allineando accuratamente sezioni adiacenti, supporta anche ricostruzioni tridimensionali di organi ed embrioni interi, trasformando pile di immagini bidimensionali in coerenti modelli 3D di espressione genica.
Cosa significa questo per gli studi tissutali futuri
Per un non specialista, INSPIRE può essere pensato come un traduttore potente che converte molte mappe geniche imperfette e rumorose in un linguaggio condiviso di schemi spaziali e programmi genici. Mantiene il contesto della posizione cellulare nel tessuto, filtra artefatti tecnici e mette in evidenza sia caratteristiche comuni sia peculiari tra gli esperimenti. Man mano che i progetti di trascrittomica spaziale si espandono fino a organi interi, tumori e organismi, metodi come INSPIRE saranno fondamentali per costruire atlanti integrati che i ricercatori potranno esplorare per comprendere come le cellule cooperano, come le malattie alterano l'architettura tissutale e come strutture complesse emergono durante lo sviluppo.
Citazione: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Parole chiave: trascrittomica spaziale, architettura tissutale, mappe di espressione genica, integrazione dei dati, microambiente tumorale