Clear Sky Science · en
Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources
Seeing Tissues as Living Maps
Biologists now have tools that read which genes are active in thousands of tiny spots across a tissue slice, turning organs, tumors and embryos into detailed molecular maps. Yet each experiment uses different instruments and setups, so the resulting maps are hard to compare or combine. This paper introduces INSPIRE, a computational method that stitches these diverse gene maps into a single, understandable view, helping scientists follow how tissues are built, change with disease and evolve over time.
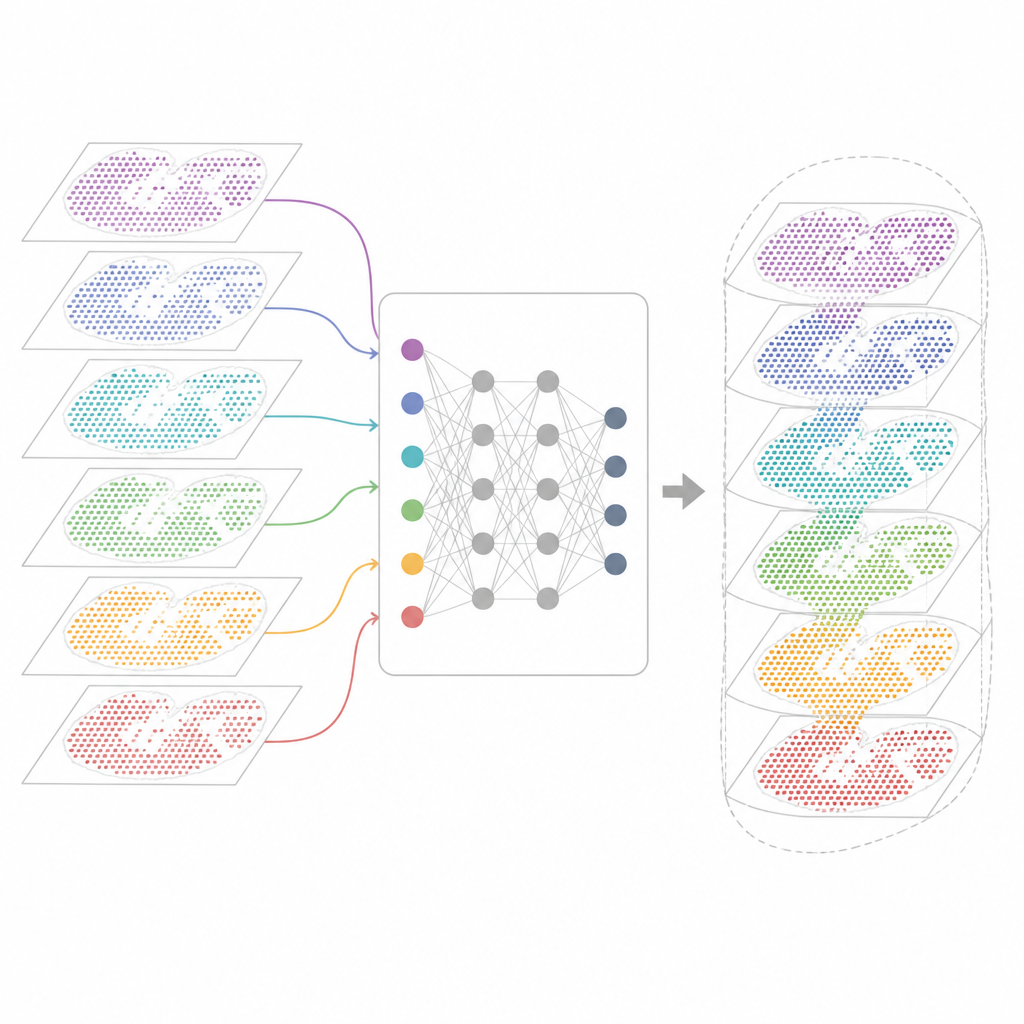
Why Many Gene Maps Are Hard to Combine
Modern spatial transcriptomics technologies measure gene activity while preserving where each cell sits in tissue. Some methods capture nearly all genes but blur together several cells in each spot. Others pinpoint single cells but only for a selected gene panel. These maps also come from different labs, machines, time points and species. As a result, each dataset carries its own technical quirks and noise. Existing analysis tools can describe one slice or a few similar slices, but they often fail when asked to align dozens of sections, bridge across technologies or keep track of both shared and unique tissue features.
A New Framework for Unifying Tissue Maps
INSPIRE tackles this integration problem with a deep learning framework that respects both the gene readouts and the physical layout of cells. It first builds a "neighborhood graph" for each tissue slice, connecting nearby spots. A graph-based neural network then transforms the raw data into a shared internal representation that mixes comparable cells from different sections while still allowing section-specific patterns to remain. An adversarial component acts like a critic, spotting where sections do not yet overlap well in this internal space and nudging the model to improve the alignment.
From Hidden Patterns to Human-Readable Features
Once the data are brought into this shared space, INSPIRE breaks the remaining signal into a set of recurring spatial patterns, called factors, each linked to a characteristic gene program. This is done using a non-negative matrix factorization step that encourages the model to represent the data as a combination of a few simple building blocks. Each factor corresponds to a spatial pattern across slices, such as a specific brain layer, a tumor niche or a developing organ region. Because INSPIRE also learns which genes are most tied to each factor, researchers can interpret these patterns in terms of known cell types and biological processes, rather than abstract numbers.
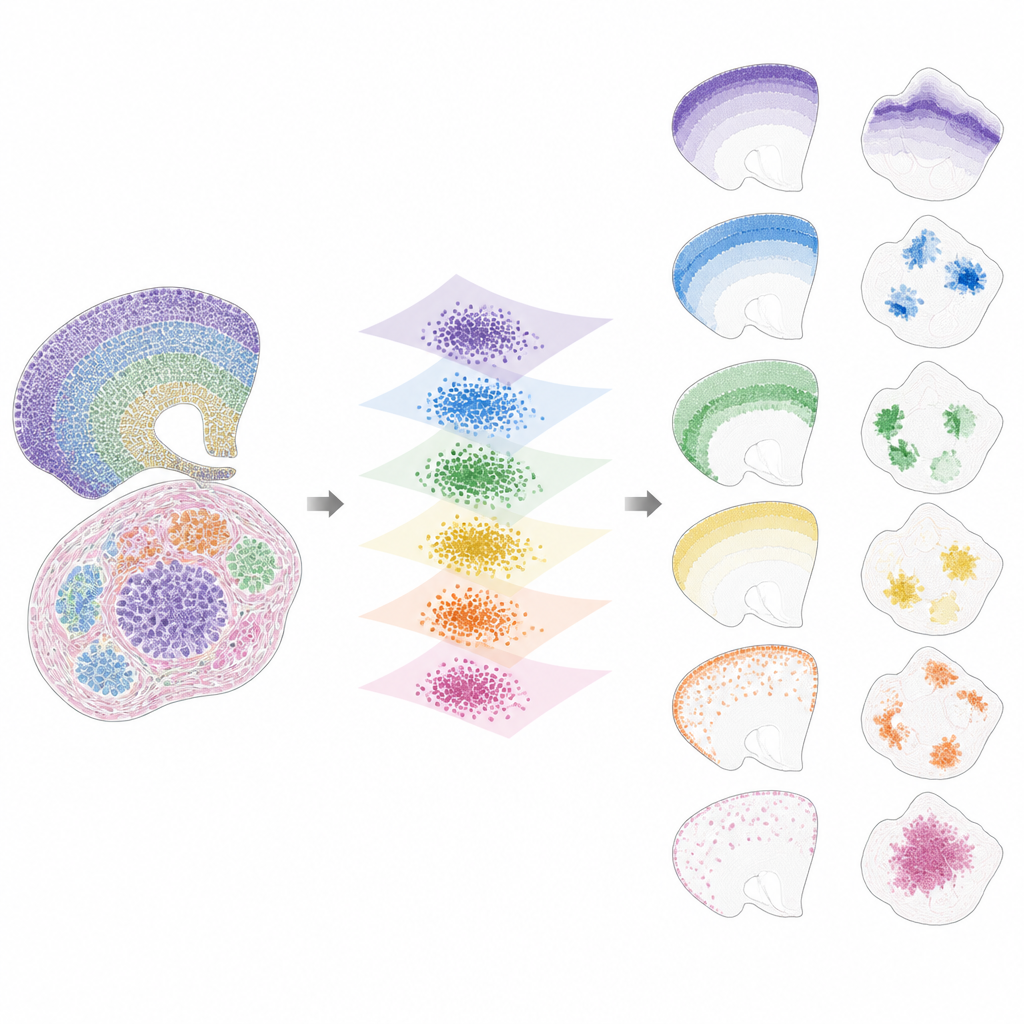
What INSPIRE Reveals in Real Datasets
The authors show that INSPIRE outperforms a range of popular methods on both simulated and real datasets. In human brain cortex slices, it aligns layers across donors and recovers fine distinctions between neuron types and supporting cells, including subtle arrangements that manual labels missed. In mouse brain, it cleanly separates shared structures, such as the cortex, from unique regions like the cerebellum, while correctly identifying their distinct gene signatures. INSPIRE also bridges different technologies, combining single-cell maps with broader surveys to transfer detailed layer information and to infer missing gene patterns that were never directly measured.
Following Disease, Development and 3D Structure
Beyond healthy tissues, INSPIRE exposes hidden variation in disease and development. In human breast cancer sections with hundreds of thousands of cells, it distinguishes noninvasive from invasive tumor regions and uncovers distinct subgroups of supporting cells around each, linked to known markers of aggressiveness and blood vessel growth. In mouse embryos, INSPIRE integrates slices taken at multiple stages to build a spatiotemporal atlas, tracking how organs such as heart, liver, lung and brain grow and reorganize. By accurately aligning adjacent slices, it also supports three-dimensional reconstructions of organs and whole embryos, turning stacks of two-dimensional images into coherent 3D gene-expression models.
What This Means for Future Tissue Studies
To a non-specialist, INSPIRE can be thought of as a powerful translator that turns many imperfect, noisy gene maps into a shared language of spatial patterns and gene programs. It keeps the context of where cells sit in tissue, filters out technical artifacts and highlights both common and unique features across experiments. As spatial transcriptomics projects scale up to whole organs, tumors and organisms, methods like INSPIRE will be key to building integrated atlases that researchers can explore to understand how cells cooperate, how diseases disrupt tissue architecture and how complex structures emerge during development.
Citation: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Keywords: spatial transcriptomics, tissue architecture, gene expression maps, data integration, tumor microenvironment