Clear Sky Science · es
Integración interpretable, flexible y con conciencia espacial de múltiples conjuntos de datos de transcriptómica espacial procedentes de fuentes diversas
Ver los tejidos como mapas vivos
Los biólogos disponen ahora de herramientas que registran qué genes están activos en miles de pequeñas localidades a lo largo de una sección tisular, transformando órganos, tumores y embriones en mapas moleculares detallados. Sin embargo, cada experimento emplea distintos instrumentos y configuraciones, por lo que los mapas resultantes son difíciles de comparar o combinar. Este artículo presenta INSPIRE, un método computacional que une estos mapas génicos diversos en una única vista comprensible, ayudando a los científicos a seguir cómo se construyen los tejidos, cómo cambian con la enfermedad y cómo evolucionan con el tiempo.
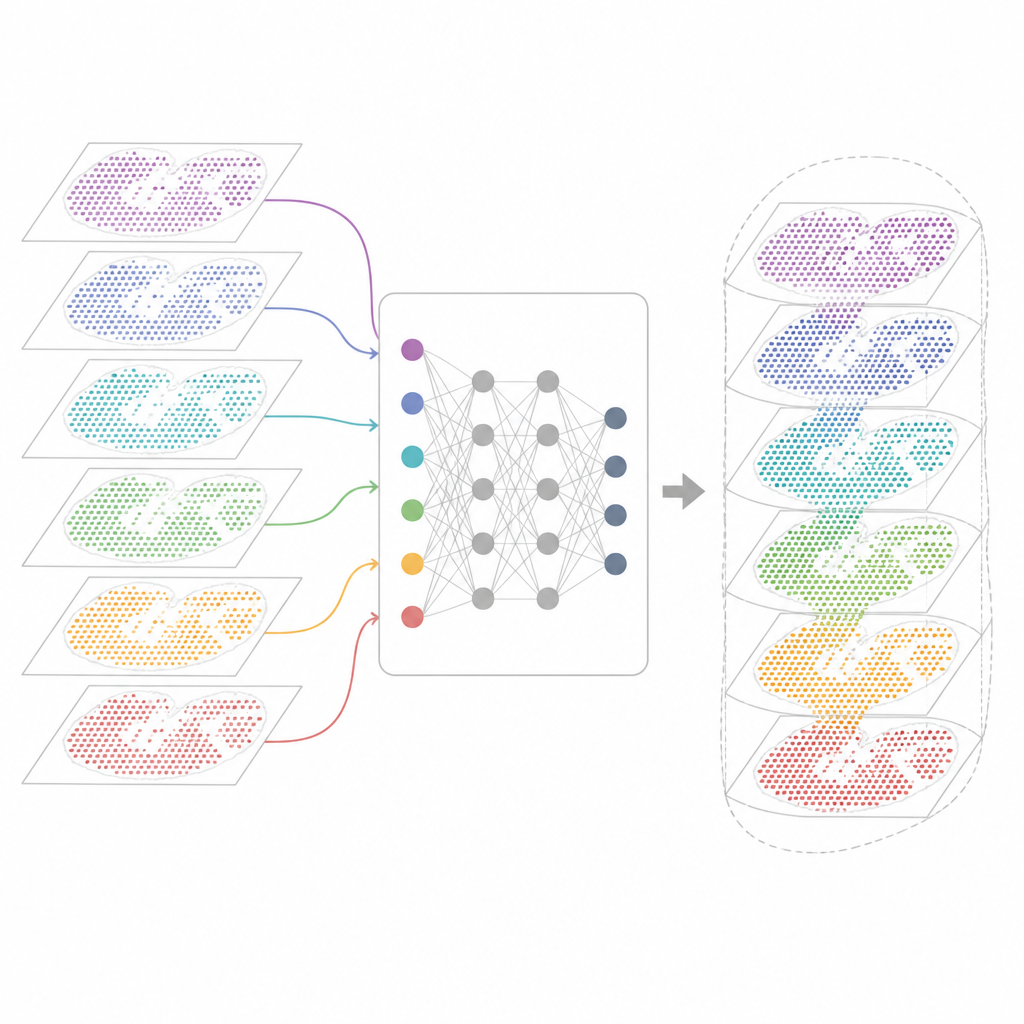
Por qué es difícil combinar muchos mapas génicos
Las tecnologías modernas de transcriptómica espacial miden la actividad génica preservando la localización de cada célula en el tejido. Algunos métodos capturan casi todos los genes pero agrupan varias células en cada punto. Otros localizan células individuales pero solo para un panel seleccionado de genes. Estos mapas también proceden de distintos laboratorios, máquinas, momentos y especies. Como resultado, cada conjunto de datos arrastra sus propias particularidades técnicas y ruido. Las herramientas de análisis existentes pueden describir una sección o unas pocas secciones similares, pero a menudo fallan cuando se les pide alinear decenas de cortes, salvar las diferencias entre tecnologías o conservar tanto las características compartidas como las específicas de cada muestra.
Un nuevo marco para unificar mapas tisulares
INSPIRE aborda este problema de integración con un marco de aprendizaje profundo que respeta tanto las lecturas génicas como la disposición física de las células. Primero construye un “grafo de vecindad” para cada sección tisular, conectando puntos cercanos. Una red neuronal basada en grafos transforma los datos crudos en una representación interna compartida que mezcla células comparables de distintas secciones mientras permite que persistan patrones específicos de cada sección. Un componente adversarial actúa como crítico, detectando dónde las secciones aún no se solapan bien en ese espacio interno e impulsando al modelo a mejorar la alineación.
De patrones ocultos a rasgos interpretables
Una vez los datos se sitúan en este espacio compartido, INSPIRE descompone la señal restante en un conjunto de patrones espaciales recurrentes, llamados factores, cada uno vinculado a un programa génico característico. Esto se realiza mediante un paso de factorización no negativa de matrices que incentiva al modelo a representar los datos como la combinación de unos pocos bloques sencillos. Cada factor corresponde a un patrón espacial a través de las secciones, como una capa concreta del cerebro, un nicho tumoral o una región en desarrollo de un órgano. Como INSPIRE también aprende qué genes están más ligados a cada factor, los investigadores pueden interpretar estos patrones en términos de tipos celulares y procesos biológicos conocidos, en lugar de números abstractos.
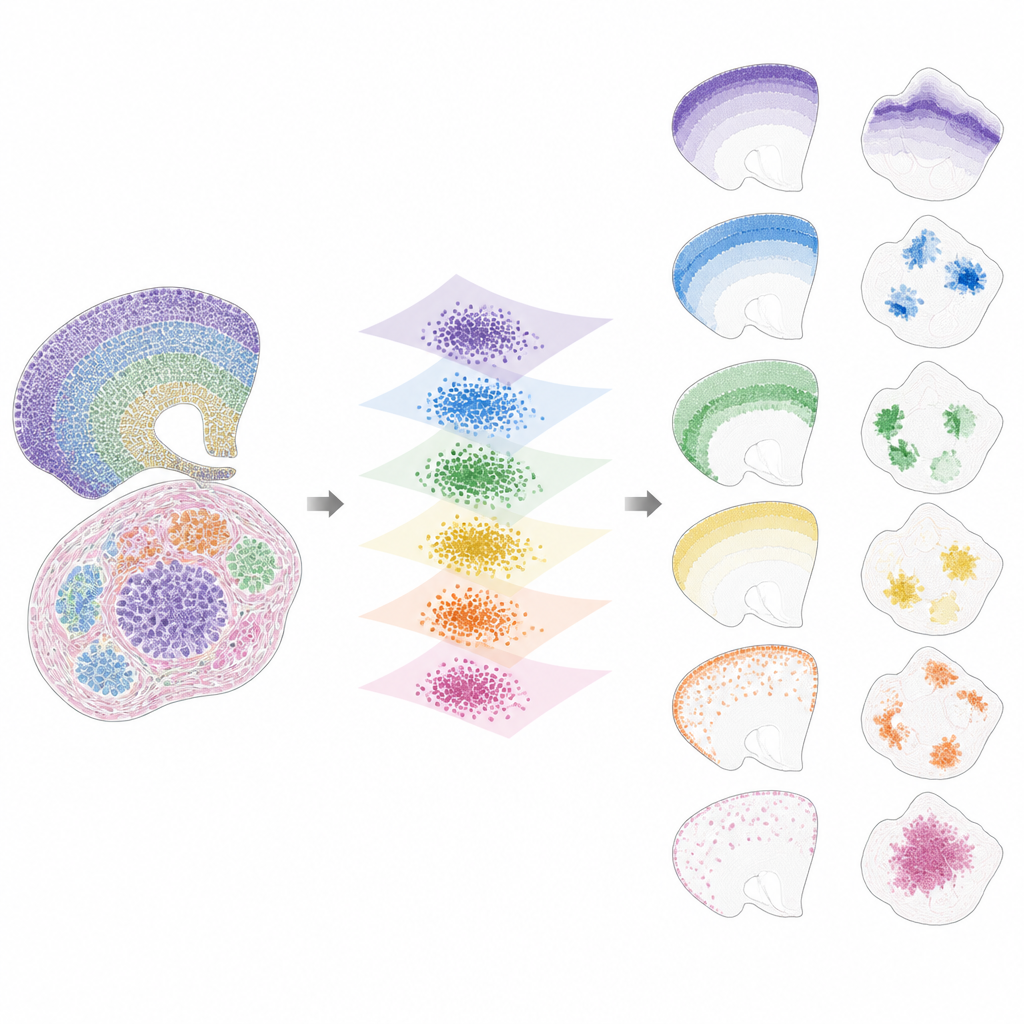
Lo que INSPIRE revela en conjuntos de datos reales
Los autores demuestran que INSPIRE supera a una variedad de métodos populares tanto en conjuntos de datos simulados como reales. En cortes de corteza cerebral humana, alinea capas entre donantes y recupera distinciones finas entre tipos de neuronas y células de soporte, incluyendo arreglos sutiles que las etiquetas manuales pasaron por alto. En cerebro de ratón, separa nítidamente estructuras compartidas, como la corteza, de regiones únicas como el cerebelo, identificando correctamente sus firmas génicas distintas. INSPIRE también salva las diferencias entre tecnologías, combinando mapas de célula única con sondeos más amplios para transferir información detallada de capas e inferir patrones génicos faltantes que nunca se midieron directamente.
Seguir la enfermedad, el desarrollo y la estructura 3D
Más allá de los tejidos sanos, INSPIRE expone variación oculta en la enfermedad y el desarrollo. En secciones de cáncer de mama humano con cientos de miles de células, distingue regiones tumorales no invasivas de invasivas y descubre subgrupos distintos de células de soporte alrededor de cada una, vinculados a marcadores conocidos de agresividad y crecimiento vascular. En embriones de ratón, INSPIRE integra cortes tomados en múltiples etapas para construir un atlas espaciotemporal, siguiendo cómo órganos como el corazón, el hígado, los pulmones y el cerebro crecen y se reorganizan. Alineando con precisión cortes adyacentes, también facilita reconstrucciones tridimensionales de órganos y embriones completos, convirtiendo pilas de imágenes bidimensionales en modelos coherentes de expresión génica en 3D.
Qué implica esto para futuros estudios tisulares
Para un no especialista, INSPIRE puede entenderse como un traductor potente que convierte muchos mapas génicos imperfectos y ruidosos en un lenguaje compartido de patrones espaciales y programas génicos. Conserva el contexto de la posición celular en el tejido, filtra artefactos técnicos y resalta rasgos tanto comunes como únicos entre experimentos. A medida que los proyectos de transcriptómica espacial se amplían a órganos, tumores y organismos enteros, métodos como INSPIRE serán clave para construir atlas integrados que los investigadores puedan explorar para entender cómo cooperan las células, cómo las enfermedades alteran la arquitectura tisular y cómo emergen estructuras complejas durante el desarrollo.
Cita: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Palabras clave: transcriptómica espacial, arquitectura tisular, mapas de expresión génica, integración de datos, microambiente tumoral