Clear Sky Science · de
Interpretierbare, flexible und räumlich bewusste Integration mehrerer Spatial-Transcriptomics-Datensätze aus verschiedenen Quellen
Gewebe als lebendige Karten sehen
Biologinnen und Biologen verfügen inzwischen über Werkzeuge, die anzeigen, welche Gene in tausenden winziger Punkte auf einem Gewebeschnitt aktiv sind, und verwandeln Organe, Tumore und Embryonen in detaillierte molekulare Karten. Jedes Experiment nutzt jedoch unterschiedliche Instrumente und Protokolle, sodass die entstehenden Karten schwer vergleichbar oder kombinierbar sind. Diese Arbeit stellt INSPIRE vor, eine rechnerische Methode, die diese vielfältigen Genkarten zu einer einzigen, verständlichen Ansicht zusammenfügt und Forschenden hilft nachzuvollziehen, wie Gewebe aufgebaut werden, sich bei Erkrankungen verändern und sich im Verlauf der Zeit entwickeln.
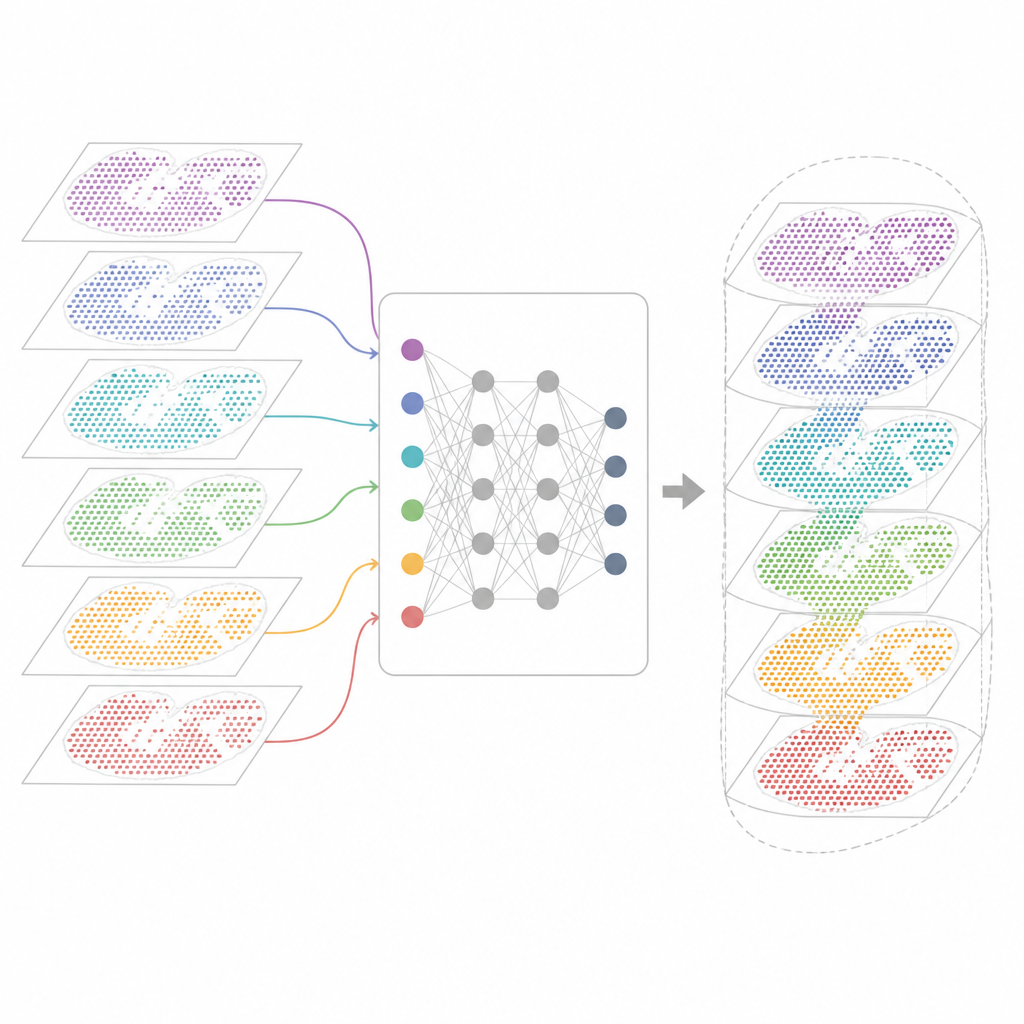
Warum viele Genkarten schwer zu kombinieren sind
Moderne Spatial‑Transcriptomics‑Technologien messen Genaktivität und erhalten gleichzeitig die räumliche Lage jeder Zelle im Gewebe. Einige Verfahren erfassen nahezu alle Gene, verwischen dabei aber mehrere Zellen in jedem Messpunkt. Andere lokalisieren einzelne Zellen, erfassen dafür aber nur ein ausgewähltes Genpanel. Diese Karten stammen zudem aus unterschiedlichen Laboren, Maschinen, Zeitpunkten und Spezies. Daher bringt jeder Datensatz seine eigenen technischen Besonderheiten und Störsignale mit. Bestehende Analysewerkzeuge können einen Schnitt oder einige ähnliche Schnitte beschreiben, scheitern jedoch oft, wenn Dutzende von Abschnitten ausgerichtet, Technologien überbrückt oder sowohl gemeinsame als auch eindeutige Gewebemerkmale berücksichtigt werden sollen.
Ein neues Rahmenwerk zur Vereinheitlichung von Gewebekarten
INSPIRE geht dieses Integrationsproblem mit einem Deep‑Learning‑Ansatz an, der sowohl die Genmessungen als auch die physische Anordnung der Zellen respektiert. Zuerst erstellt es für jeden Gewebeschnitt einen "Nachbarschaftsgraphen", der benachbarte Messpunkte verbindet. Ein graphbasiertes neuronales Netz transformiert dann die Rohdaten in eine gemeinsame interne Repräsentation, die vergleichbare Zellen aus verschiedenen Abschnitten zusammenführt, gleichzeitig aber abschnittsspezifische Muster erhalten lässt. Eine adversariale Komponente fungiert wie ein Kritiker, erkennt Bereiche, in denen Abschnitte in diesem internen Raum noch nicht gut überlappen, und veranlasst das Modell, die Ausrichtung zu verbessern.
Von verborgenen Mustern zu menschenlesbaren Merkmalen
Sobald die Daten in diesen gemeinsamen Raum überführt sind, zerlegt INSPIRE das verbleibende Signal in eine Menge wiederkehrender räumlicher Muster, sogenannte Faktoren, von denen jeder mit einem charakteristischen Genprogramm verknüpft ist. Dies geschieht mittels einer nichtnegativen Matrixfaktorisierung, die das Modell dazu anregt, die Daten als Kombination weniger einfacher Bausteine darzustellen. Jeder Faktor entspricht einem räumlichen Muster über Schnitte hinweg, etwa einer bestimmten Hirnschicht, einer Tumornische oder einem Bereich eines sich entwickelnden Organs. Da INSPIRE außerdem lernt, welche Gene am stärksten mit jedem Faktor assoziiert sind, können Forschende diese Muster in Bezug auf bekannte Zelltypen und biologische Prozesse interpretieren statt als abstrakte Zahlen.
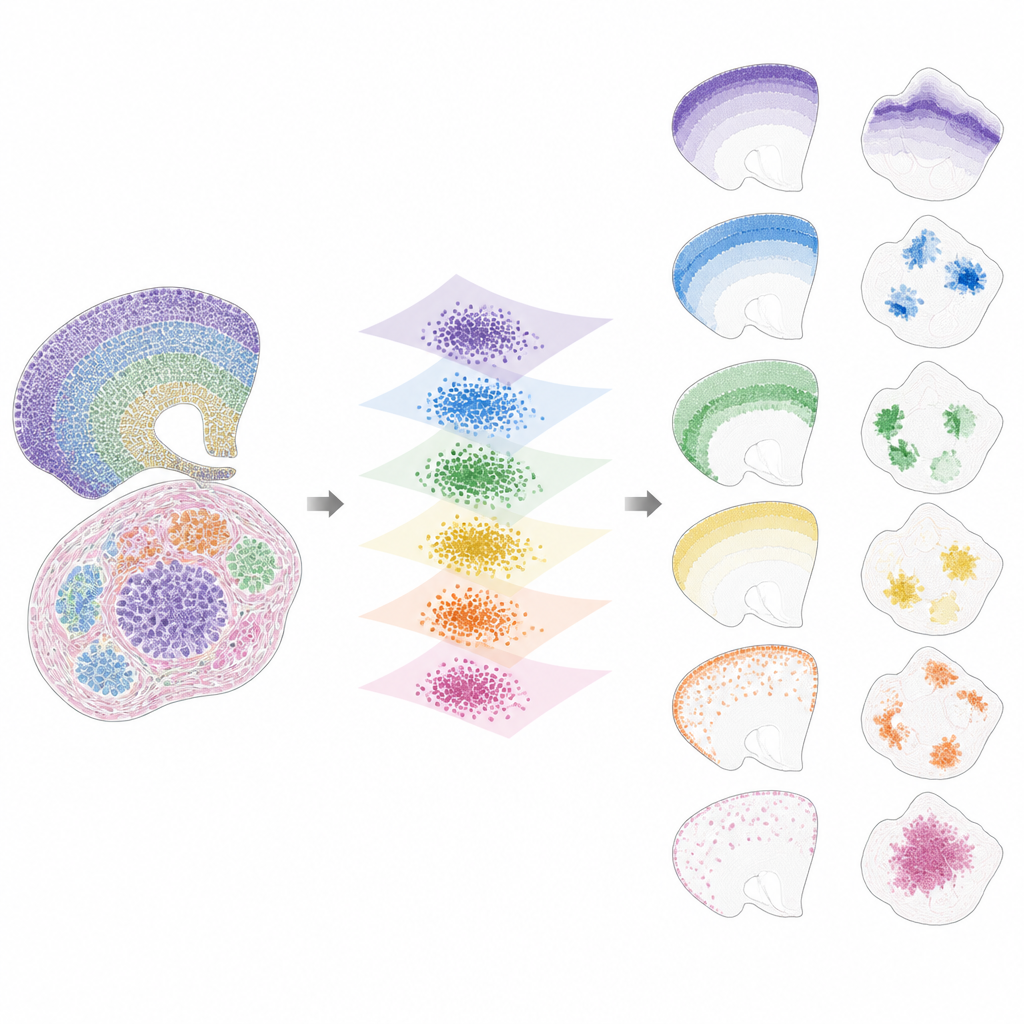
Was INSPIRE in echten Datensätzen enthüllt
Die Autorinnen und Autoren zeigen, dass INSPIRE eine Reihe gängiger Methoden sowohl auf simulierten als auch auf realen Datensätzen übertrifft. In menschlichen Kortex‑Schnitten richtet es Schichten über Spender hinweg aus und erkennt feine Unterschiede zwischen Neuronen‑Typen und Stütz‑Zellen, einschließlich subtiler Anordnungen, die manuelle Labels verpasst hatten. Im Mausgehirn trennt es sauber gemeinsame Strukturen wie den Kortex von einzigartigen Regionen wie dem Kleinhirn und identifiziert korrekt deren unterschiedliche Gensignaturen. INSPIRE überbrückt zudem verschiedene Technologien, kombiniert Single‑Cell‑Karten mit breiteren Übersichten, um detaillierte Schichtinformationen zu übertragen und fehlende Genmuster zu erschließen, die nie direkt gemessen wurden.
Krankheit, Entwicklung und 3D‑Struktur verfolgen
Über gesunde Gewebe hinaus legt INSPIRE verborgene Variationen in Krankheit und Entwicklung offen. In menschlichen Brustkrebs‑Schnitten mit Hunderttausenden von Zellen unterscheidet es nichtinvasive von invasiven Tumorregionen und deckt unterschiedliche Untergruppen von Stütz‑Zellen in deren Umgebung auf, die mit bekannten Markern für Aggressivität und Gefäßneubildung verknüpft sind. In Maus‑Embryonen integriert INSPIRE Schnitte aus mehreren Stadien, um ein raum‑zeitliches Atlasbild zu erstellen und nachzuvollziehen, wie sich Organe wie Herz, Leber, Lunge und Gehirn vergrößern und umorganisieren. Durch die genaue Ausrichtung benachbarter Schnitte unterstützt es auch dreidimensionale Rekonstruktionen von Organen und gesamten Embryonen und wandelt Stapel zweidimensionaler Bilder in kohärente 3D‑Modelle der Genexpression um.
Was das für künftige Gewebestudien bedeutet
Für Nicht‑Spezialisten lässt sich INSPIRE als leistungsfähiger Übersetzer begreifen, der viele unvollkommene, verrauschte Genkarten in eine gemeinsame Sprache aus räumlichen Mustern und Genprogrammen überführt. Es bewahrt den Kontext, wo Zellen im Gewebe sitzen, filtert technische Artefakte heraus und hebt sowohl gemeinsame als auch einzigartige Merkmale über Experimente hinweg hervor. Wenn Spatial‑Transcriptomics‑Projekte auf ganze Organe, Tumore und Organismen skaliert werden, werden Methoden wie INSPIRE entscheidend sein, um integrierte Atlanten zu erstellen, die Forschende nutzen können, um zu verstehen, wie Zellen zusammenarbeiten, wie Krankheiten Gewebearchitektur stören und wie komplexe Strukturen während der Entwicklung entstehen.
Zitation: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Schlüsselwörter: spatial transcriptomics, Gewebearchitektur, Karten der Genexpression, Datenintegration, Tumormikroumgebung