Clear Sky Science · fr
Intégration interprétable, flexible et spatiale de multiples jeux de données de transcriptomique spatiale issus de sources diverses
Voir les tissus comme des cartes vivantes
Les biologistes disposent aujourd'hui d'outils qui lisent quels gènes sont actifs sur des milliers de petits points d'une coupe tissulaire, transformant organes, tumeurs et embryons en cartes moléculaires détaillées. Pourtant, chaque expérience utilise des instruments et des protocoles différents, ce qui rend ces cartes difficiles à comparer ou à combiner. Cet article présente INSPIRE, une méthode computationnelle qui assemble ces cartes géniques hétérogènes en une vue unique et compréhensible, aidant les chercheurs à suivre comment les tissus se construisent, évoluent avec la maladie et se modifient au fil du temps.
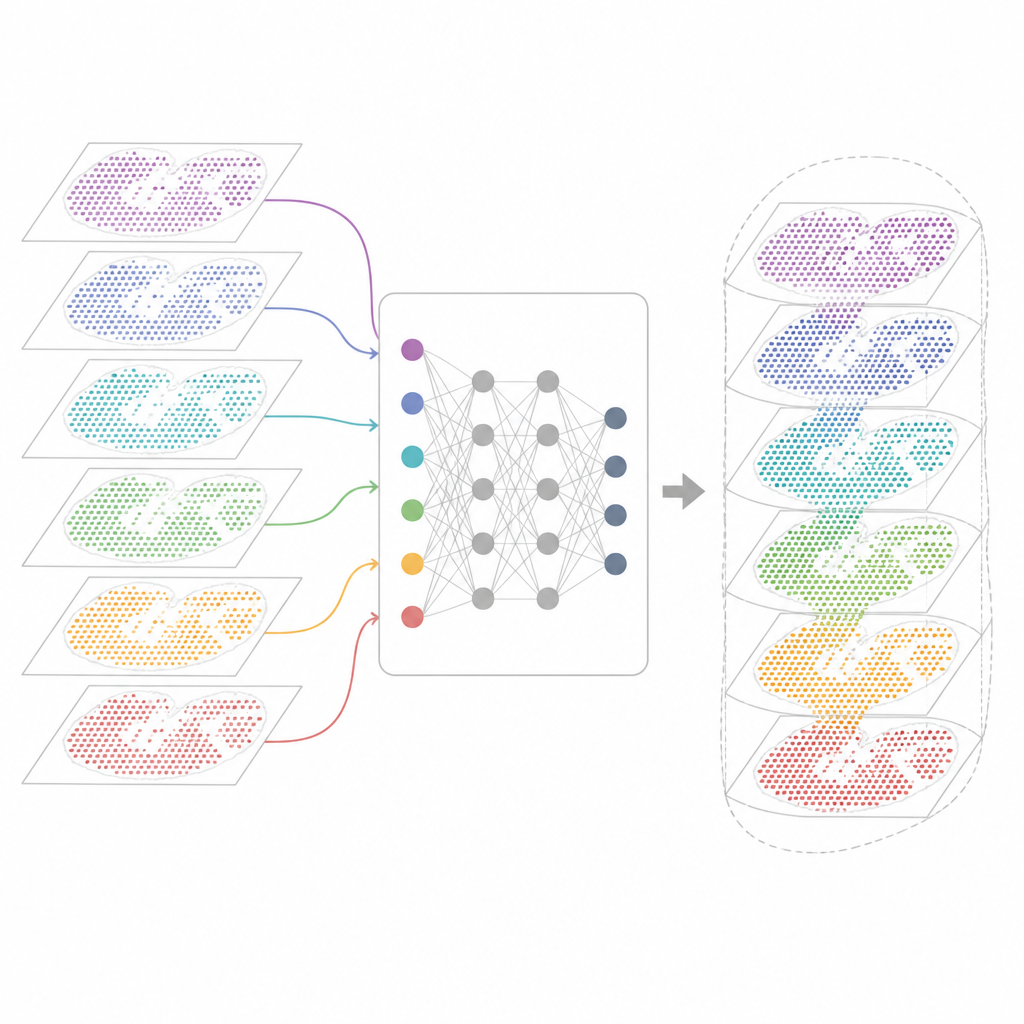
Pourquoi de nombreuses cartes géniques sont difficiles à combiner
Les technologies modernes de transcriptomique spatiale mesurent l'activité génique tout en préservant la position de chaque cellule dans le tissu. Certaines méthodes capturent presque tous les gènes mais brouillent plusieurs cellules dans chaque point. D'autres localisent des cellules individuelles mais seulement pour un panel de gènes sélectionnés. Ces cartes proviennent aussi de différents laboratoires, machines, moments et espèces. En conséquence, chaque jeu de données porte ses propres artefacts techniques et du bruit. Les outils d'analyse existants peuvent décrire une coupe ou quelques coupes similaires, mais ils échouent souvent lorsqu'on leur demande d'aligner des dizaines de sections, de faire le lien entre des technologies ou de conserver à la fois les caractéristiques partagées et spécifiques des tissus.
Un nouveau cadre pour unifier les cartes tissulaires
INSPIRE aborde ce problème d'intégration avec un cadre d'apprentissage profond qui respecte à la fois les mesures géniques et la disposition physique des cellules. Il construit d'abord un « graphe de voisinage » pour chaque coupe tissulaire, reliant les points voisins. Un réseau de neurones basé sur des graphes transforme ensuite les données brutes en une représentation interne partagée qui mélange les cellules comparables provenant de différentes sections tout en permettant aux motifs spécifiques à une section de subsister. Un composant antagoniste joue le rôle de critique, repérant les endroits où les sections ne se recouvrent pas encore bien dans cet espace interne et poussant le modèle à améliorer l'alignement.
Des motifs cachés vers des caractéristiques interprétables
Une fois les données projetées dans cet espace partagé, INSPIRE décompose le signal restant en un ensemble de motifs spatiaux récurrents, appelés facteurs, chacun lié à un programme génique caractéristique. Cela se fait via une étape de factorisation en matrices non négatives qui encourage le modèle à représenter les données comme la combinaison d'un petit nombre de blocs simples. Chaque facteur correspond à un motif spatial à travers les coupes, comme une couche cérébrale spécifique, une niche tumorale ou une région d'organe en développement. Parce qu'INSPIRE apprend aussi quels gènes sont le plus associés à chaque facteur, les chercheurs peuvent interpréter ces motifs en termes de types cellulaires et de processus biologiques connus, plutôt qu'en chiffres abstraits.
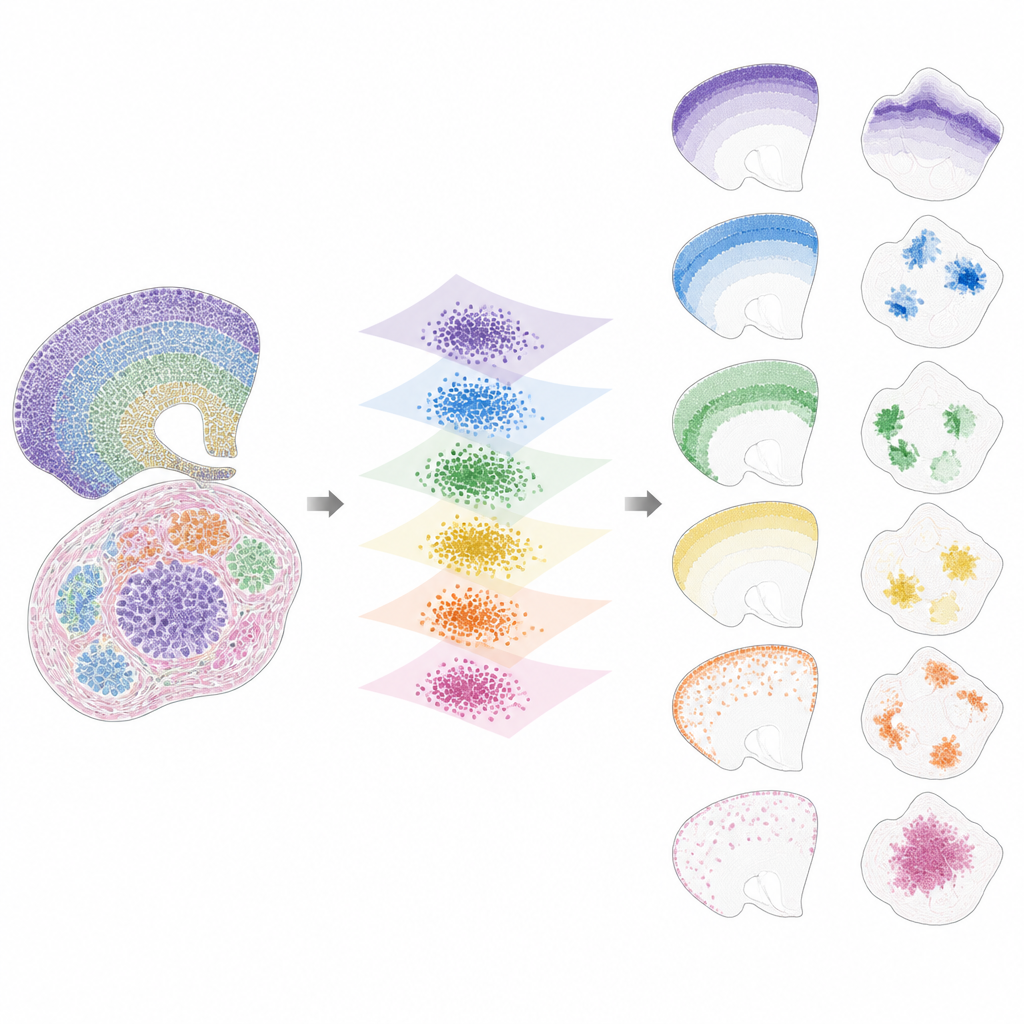
Ce qu'INSPIRE révèle dans des jeux de données réels
Les auteurs montrent qu'INSPIRE surpasse une gamme de méthodes populaires sur des jeux de données simulés et réels. Dans des coupes de cortex humain, il aligne les couches entre donneurs et retrouve des distinctions fines entre types de neurones et cellules de soutien, y compris des agencements subtils manqués par l'annotation manuelle. Chez la souris, il sépare clairement les structures partagées, comme le cortex, des régions uniques comme le cervelet, tout en identifiant correctement leurs signatures géniques distinctes. INSPIRE relie également différentes technologies, combinant des cartes monocellulaires avec des relevés plus larges pour transférer des informations de couche détaillées et inférer des motifs géniques manquants qui n'ont jamais été mesurés directement.
Suivre la maladie, le développement et la structure 3D
Au-delà des tissus sains, INSPIRE met en évidence des variations cachées dans la maladie et le développement. Dans des sections de cancer du sein humain contenant des centaines de milliers de cellules, il distingue les régions tumorales non invasives des régions invasives et dévoile des sous-groupes distincts de cellules de soutien autour de chacune, liés à des marqueurs connus d'agressivité et d'angiogenèse. Chez l'embryon de souris, INSPIRE intègre des coupes prises à plusieurs stades pour construire un atlas spatiotemporel, suivant comment des organes tels que le cœur, le foie, les poumons et le cerveau grandissent et se réorganisent. En alignant avec précision des coupes adjacentes, il facilite aussi des reconstructions tridimensionnelles d'organes et d'embryons entiers, transformant des empilements d'images bidimensionnelles en modèles 3D cohérents d'expression génique.
Ce que cela signifie pour les études tissulaires futures
Pour un non-spécialiste, INSPIRE peut être vu comme un puissant traducteur qui transforme de nombreuses cartes géniques imparfaites et bruyantes en un langage partagé de motifs spatiaux et de programmes géniques. Il conserve le contexte de la position des cellules dans le tissu, filtre les artefacts techniques et met en lumière les caractéristiques communes et uniques entre expériences. À mesure que les projets de transcriptomique spatiale s'étendent à des organes entiers, des tumeurs et des organismes complets, des méthodes comme INSPIRE seront essentielles pour construire des atlas intégrés que les chercheurs pourront explorer afin de comprendre comment les cellules coopèrent, comment les maladies perturbent l'architecture tissulaire et comment des structures complexes émergent au cours du développement.
Citation: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Mots-clés: transcriptomique spatiale, architecture tissulaire, cartes d'expression génique, intégration de données, microenvironnement tumoral