Clear Sky Science · pl
Interpretowalna, elastyczna i uwzględniająca przestrzeń integracja wielu zestawów danych z przestrzennej transkryptomiki pochodzących z różnych źródeł
Postrzeganie tkanek jako żywych map
Biolodzy mają dziś narzędzia, które odczytują, które geny są aktywne w tysiącach drobnych punktów na wycinku tkanki, zamieniając organy, guzy i embriony w szczegółowe mapy molekularne. Jednak każde doświadczenie używa innych instrumentów i ustawień, więc powstałe mapy trudno porównać lub połączyć. W artykule przedstawiono INSPIRE, metodę obliczeniową, która zszywa te różnorodne mapy genowe w jeden zrozumiały widok, pomagając naukowcom śledzić, jak tkanki są zbudowane, jak zmieniają się przy chorobie i jak ewoluują w czasie.
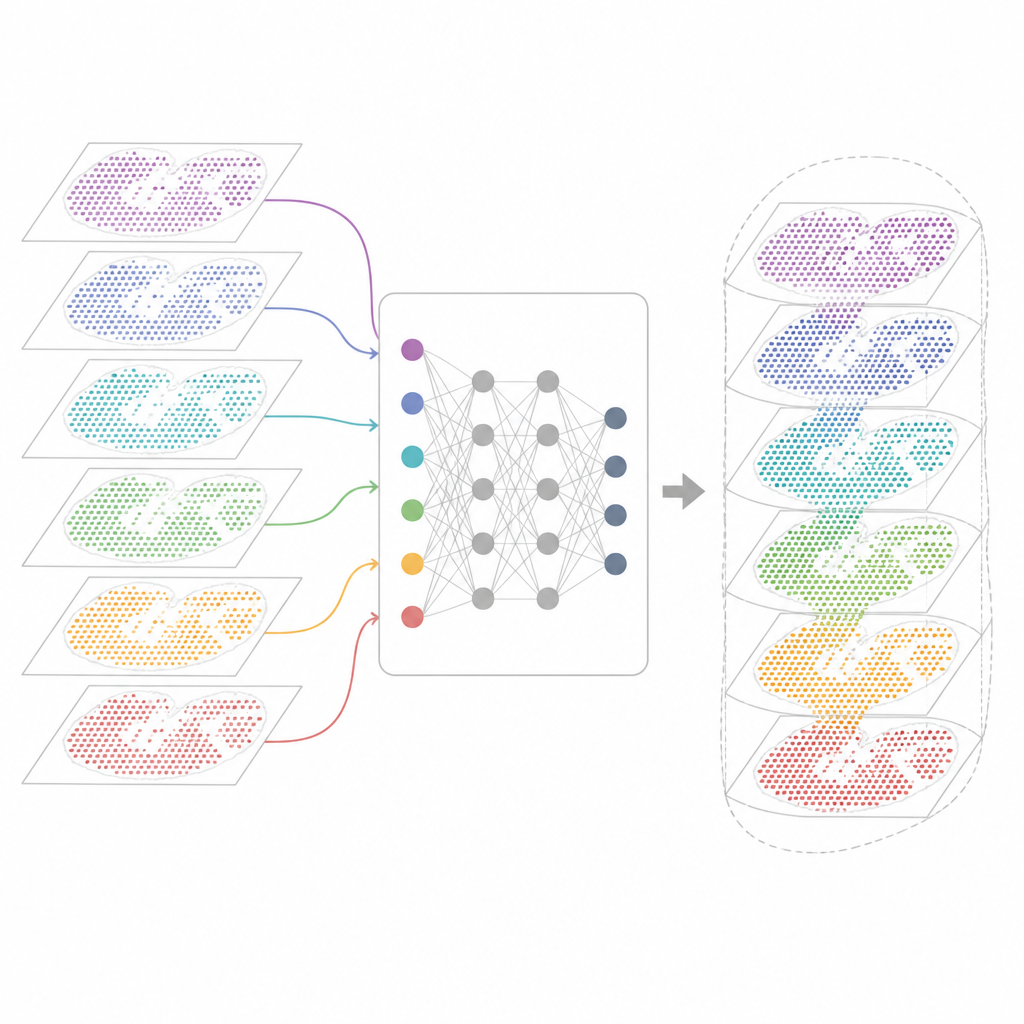
Dlaczego wiele map genowych trudno połączyć
Nowoczesne technologie przestrzennej transkryptomiki mierzą aktywność genów, zachowując informację o położeniu każdej komórki w tkance. Niektóre metody wychwytują niemal wszystkie geny, ale zlewają w jednym punkcie sygnały kilku komórek. Inne precyzyjnie lokalizują pojedyncze komórki, lecz tylko w wyselekcjonowanym panelu genów. Mapy te pochodzą też z różnych laboratoriów, maszyn, punktów czasowych i gatunków. W rezultacie każdy zestaw danych niesie własne techniczne cechy i szumy. Istniejące narzędzia analityczne mogą opisywać pojedynczy wycinek lub kilka podobnych wycinków, ale często zawodzą, gdy trzeba wyrównać dziesiątki sekcji, połączyć różne technologie lub jednocześnie zachować cechy wspólne i specyficzne dla poszczególnych zestawów.
Nowe ramy do jednoczenia map tkanek
INSPIRE podchodzi do problemu integracji za pomocą ram uczenia głębokiego, które uwzględniają zarówno odczyty genów, jak i fizyczne rozmieszczenie komórek. Najpierw buduje „graf sąsiedztwa” dla każdego wycinka tkanki, łącząc pobliskie punkty. Następnie sieć neuronowa oparta na grafach przekształca surowe dane w wspólną reprezentację wewnętrzną, która miesza porównywalne komórki z różnych sekcji, jednocześnie pozwalając zachować wzory charakterystyczne dla poszczególnych sekcji. Składnik adwersarialny pełni rolę krytyka, wykrywając obszary, gdzie sekcje jeszcze się nie pokrywają w tej przestrzeni wewnętrznej, i popycha model do poprawy wyrównania.
Od ukrytych wzorów do czytelnych cech
Po sprowadzeniu danych do wspólnej przestrzeni INSPIRE rozkłada pozostały sygnał na zestaw powtarzających się wzorów przestrzennych, zwanych czynnikami, z których każdy powiązany jest z charakterystycznym programem genowym. Odbywa się to za pomocą kroku niedodatniej faktoryzacji macierzy, który skłania model do reprezentowania danych jako kombinacji kilku prostych bloków budulcowych. Każdy czynnik odpowiada wzorowi przestrzennemu występującemu w przekrojach, takim jak konkretna warstwa mózgu, nisza nowotworowa czy obszar rozwijającego się narządu. Ponieważ INSPIRE uczy się też, które geny są najsilniej powiązane z każdym czynnikiem, badacze mogą interpretować te wzory w kategoriach znanych typów komórek i procesów biologicznych, zamiast abstrakcyjnych liczb.
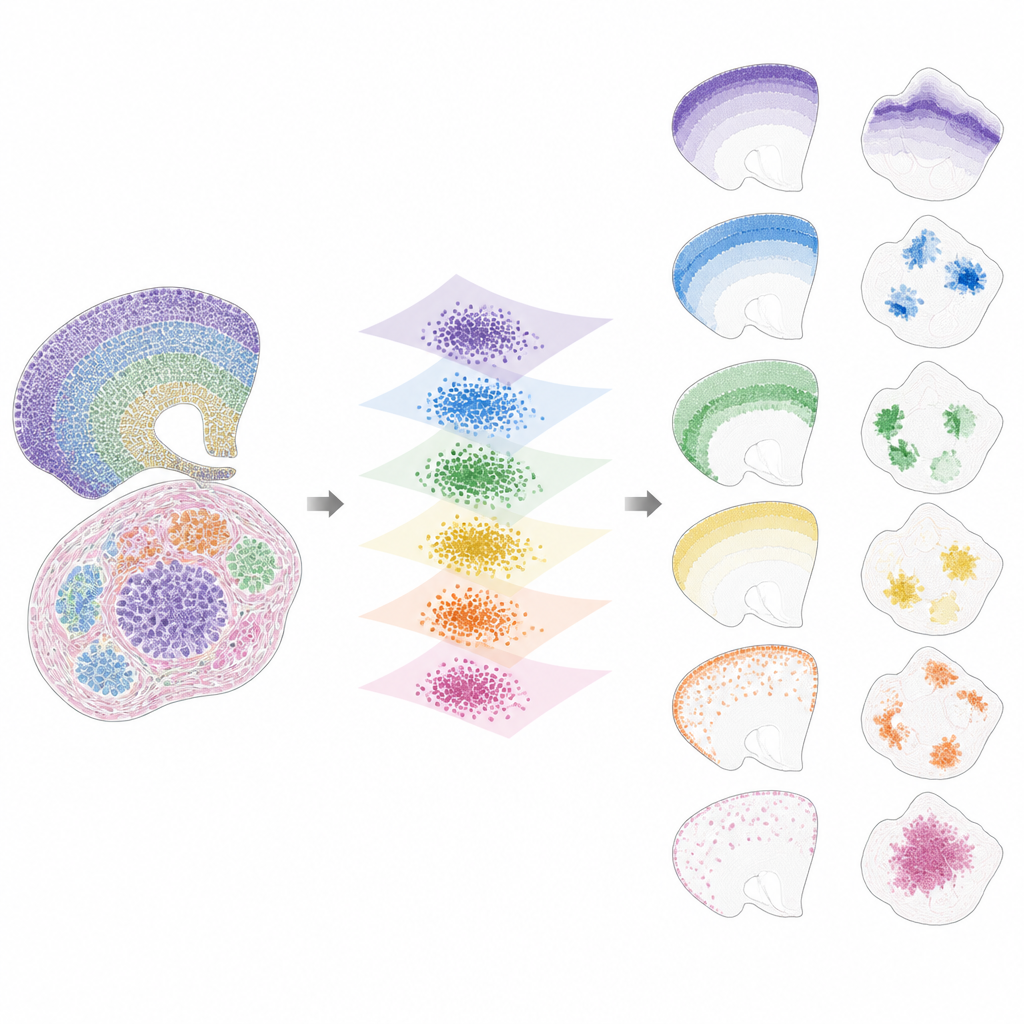
Co INSPIRE ujawnia na rzeczywistych zestawach danych
Autorzy pokazują, że INSPIRE przewyższa szereg popularnych metod zarówno na danych symulowanych, jak i rzeczywistych. W przekrojach kory mózgowej człowieka wyrównuje warstwy między dawcami i odzyskuje subtelne rozróżnienia między typami neuronów i komórek wspierających, w tym układy, które umknęły etykietom ręcznym. W mózgu myszy wyraźnie oddziela struktury wspólne, takie jak kora, od unikatowych regionów jak móżdżek, prawidłowo identyfikując ich odrębne sygnatury genowe. INSPIRE łączy również różne technologie, scalając mapy pojedynczych komórek z szerszymi przeglądami, by przenosić szczegółowe informacje o warstwach i wnioskować brakujące wzory genowe, które nigdy nie były mierzone bezpośrednio.
Śledzenie chorób, rozwoju i struktury 3D
Poza tkankami zdrowymi, INSPIRE ujawnia ukrytą zmienność w chorobie i rozwoju. W przekrojach ludzkiego raka piersi zawierających setki tysięcy komórek rozróżnia obszary nowotworu nieinwazyjnego od inwazyjnego i odkrywa odrębne podgrupy komórek wspierających wokół każdego z nich, powiązane ze znanymi markerami agresywności i angiogenezy. U zarodków myszy INSPIRE integruje wycinki pobrane na różnych etapach, budując atlas czasoprzestrzenny i śledząc, jak narządy takie jak serce, wątroba, płuca i mózg rosną i reorganizują się. Poprzez dokładne wyrównanie sąsiednich sekcji wspiera także rekonstrukcje trójwymiarowe narządów i całych zarodków, przekształcając stosy obrazów dwuwymiarowych w spójne modele 3D ekspresji genów.
Co to oznacza dla przyszłych badań nad tkankami
Dla laika INSPIRE można uznać za potężnego tłumacza, który przekształca wiele niedoskonałych, zaszumionych map genowych w wspólny język wzorów przestrzennych i programów genowych. Zachowuje kontekst położenia komórek w tkance, filtruje artefakty techniczne i uwypukla zarówno cechy wspólne, jak i unikatowe między eksperymentami. W miarę jak projekty przestrzennej transkryptomiki skalują się do całych organów, guzów i organizmów, metody takie jak INSPIRE będą kluczowe do budowy zintegrowanych atlasów, które badacze mogą eksplorować, by zrozumieć, jak komórki współpracują, jak choroby zakłócają architekturę tkanek i jak złożone struktury powstają podczas rozwoju.
Cytowanie: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Słowa kluczowe: przestrzenna transkryptomika, architektura tkanki, mapy ekspresji genów, integracja danych, mikrośrodowisko guza