Clear Sky Science · nl
Interpreteerbare, flexibele en ruimtelijk-bewuste integratie van meerdere spatial-transcriptomics datasets uit diverse bronnen
Weefsels zien als levende kaarten
Biologen beschikken nu over instrumenten die vastleggen welke genen actief zijn op duizenden kleine plekjes verspreid over een weefseldunne plak, waardoor organen, tumoren en embryo's veranderen in gedetailleerde moleculaire kaarten. Elk experiment gebruikt echter verschillende apparaten en opstellingen, waardoor de resulterende kaarten moeilijk te vergelijken of te combineren zijn. Dit artikel introduceert INSPIRE, een computationele methode die deze uiteenlopende genkaarten aan elkaar naait tot één begrijpelijk geheel, en onderzoekers helpt te volgen hoe weefsels zijn opgebouwd, veranderen bij ziekte en zich in de tijd ontwikkelen.
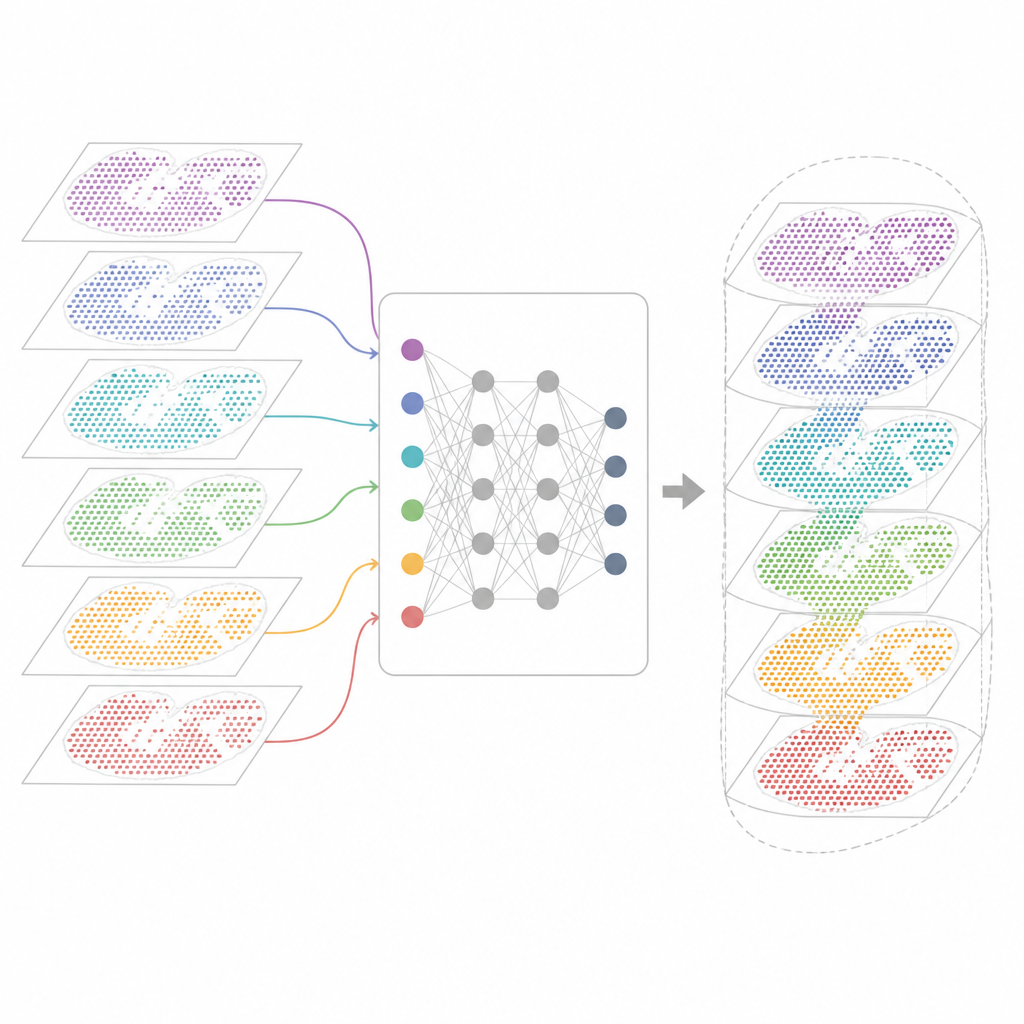
Waarom veel genkaarten moeilijk te combineren zijn
Moderne spatial-transcriptomics-technologieën meten genactiviteit terwijl ze de fysieke positie van cellen in het weefsel behouden. Sommige methoden leggen bijna alle genen vast, maar vervagen meerdere cellen in elk meetpunt. Andere pinpointen individuele cellen, maar alleen voor een beperkte genpanel. Deze kaarten komen ook uit verschillende laboratoria, apparaten, tijdstippen en soorten. Als gevolg draagt elk dataset zijn eigen technische eigenaardigheden en ruis. Bestaande analysetools kunnen één plak of een paar vergelijkbare plakken beschrijven, maar falen vaak wanneer ze tientallen secties moeten afstemmen, technologieën moeten overbruggen of zowel gedeelde als unieke weefselkenmerken moeten vastleggen.
Een nieuw kader om weefselkaarten te verenigen
INSPIRE pakt dit integratieprobleem aan met een deep-learningkader dat zowel de genmetingen als de fysieke indeling van cellen respecteert. Het bouwt eerst een "buurtgrafiek" voor elke weefselsectie, waarmee nabijgelegen meetpunten worden verbonden. Een grafiekgebaseerd neuraal netwerk transformeert vervolgens de ruwe data naar een gedeelde interne representatie die vergelijkbare cellen uit verschillende secties samenbrengt, terwijl sectiespecifieke patronen behouden kunnen blijven. Een adversariële component fungeert als criticus die plekken opmerkt waar secties in deze interne ruimte nog niet goed overlappen en het model aanstuurt om de uitlijning te verbeteren.
Van verborgen patronen naar mensleesbare kenmerken
Zodra de data in deze gedeelde ruimte zijn gebracht, splitst INSPIRE het resterende signaal op in een set terugkerende ruimtelijke patronen, factoren genoemd, die elk gekoppeld zijn aan een karakteristiek genprogramma. Dit gebeurt met een non-negatieve matrixfactorisatiestap die het model aanmoedigt de data te representeren als een combinatie van enkele eenvoudige bouwstenen. Elke factor komt overeen met een ruimtelijk patroon over secties, zoals een specifieke hersenlaag, een tumorniche of een regio in ontwikkeling. Omdat INSPIRE ook leert welke genen het meest aan elke factor verbonden zijn, kunnen onderzoekers deze patronen interpreteren in termen van bekende celtypen en biologische processen, in plaats van als abstracte getallen.
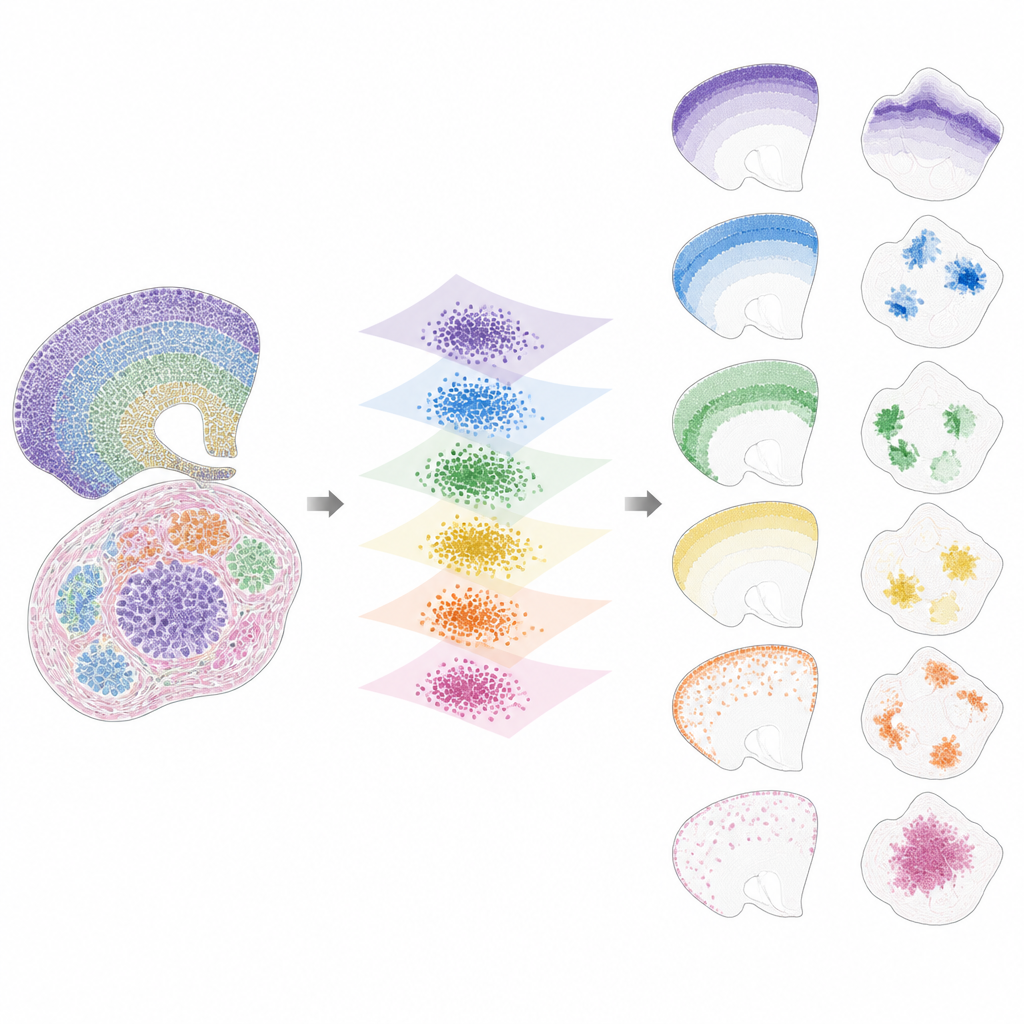
Wat INSPIRE onthult in echte datasets
De auteurs tonen aan dat INSPIRE beter presteert dan een reeks populaire methoden op zowel gesimuleerde als echte datasets. In menselijke cortexplakken lijnt het lagen over donors uit en herstelt het fijne onderscheidingen tussen neurontypen en ondersteunende cellen, inclusief subtiele ordeningen die handmatige labels misten. In muizenhersenen scheidt het duidelijk gedeelde structuren, zoals de cortex, van unieke regio's zoals het cerebellum, terwijl het hun onderscheidende gensignaturen correct identificeert. INSPIRE overbrugt ook verschillende technologieën, door single-cell-kaarten te combineren met ruimere onderzoeken om gedetailleerde laaginformatie over te dragen en ontbrekende genpatronen af te leiden die nooit direct werden gemeten.
Ziekte, ontwikkeling en 3D-structuur volgen
Buiten gezonde weefsels onthult INSPIRE verborgen variatie in ziekte en ontwikkeling. In menselijke borstkankerplakken met honderdduizenden cellen onderscheidt het niet-invasieve van invasieve tumorregio's en ontdekt het verschillende subgroepen van ondersteunende cellen rondom elk, gekoppeld aan bekende markers van agressiviteit en bloedvatvorming. In muisembryo's integreert INSPIRE plakken die op meerdere stadia zijn genomen om een spatiotemporale atlas te bouwen, waarmee te volgen is hoe organen zoals hart, lever, long en hersenen groeien en zich reorganiseren. Door aangrenzende plakken nauwkeurig uit te lijnen ondersteunt het ook driedimensionale reconstructies van organen en hele embryo's, waarmee stapels tweedimensionale beelden worden omgezet in coherente 3D-modellen van genexpressie.
Wat dit betekent voor toekomstige weefselstudies
Voor niet-specialisten kan INSPIRE worden gezien als een krachtige vertaler die vele onvolmaakte, rumoerige genkaarten omzet in een gedeelde taal van ruimtelijke patronen en genprogramma's. Het behoudt de context van waar cellen in het weefsel liggen, filtert technische artefacten weg en benadrukt zowel gemeenschappelijke als unieke kenmerken tussen experimenten. Naarmate spatial-transcriptomics-projecten opschalen naar gehele organen, tumoren en organismen, zullen methoden zoals INSPIRE cruciaal zijn voor het bouwen van geïntegreerde atlassen die onderzoekers kunnen verkennen om te begrijpen hoe cellen samenwerken, hoe ziekten de weefselarchitectuur ontregelen en hoe complexe structuren zich tijdens ontwikkeling vormen.
Bronvermelding: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Trefwoorden: spatial transcriptomics, weefselarchitectuur, genexpressiekaarten, data-integratie, tumormicro-omgeving