Clear Sky Science · pt
Integração interpretável, flexível e espacialmente consciente de múltiplos conjuntos de dados de transcriptômica espacial de fontes diversas
Ver Tecidos como Mapas Vivos
Os biólogos hoje dispõem de ferramentas que leem quais genes estão ativos em milhares de pequenos pontos ao longo de uma lâmina de tecido, transformando órgãos, tumores e embriões em mapas moleculares detalhados. Ainda assim, cada experimento usa instrumentos e configurações diferentes, de modo que os mapas resultantes são difíceis de comparar ou combinar. Este artigo apresenta o INSPIRE, um método computacional que costura esses diversos mapas gênicos em uma única visão compreensível, ajudando os cientistas a acompanhar como os tecidos são formados, como mudam com a doença e como evoluem ao longo do tempo.
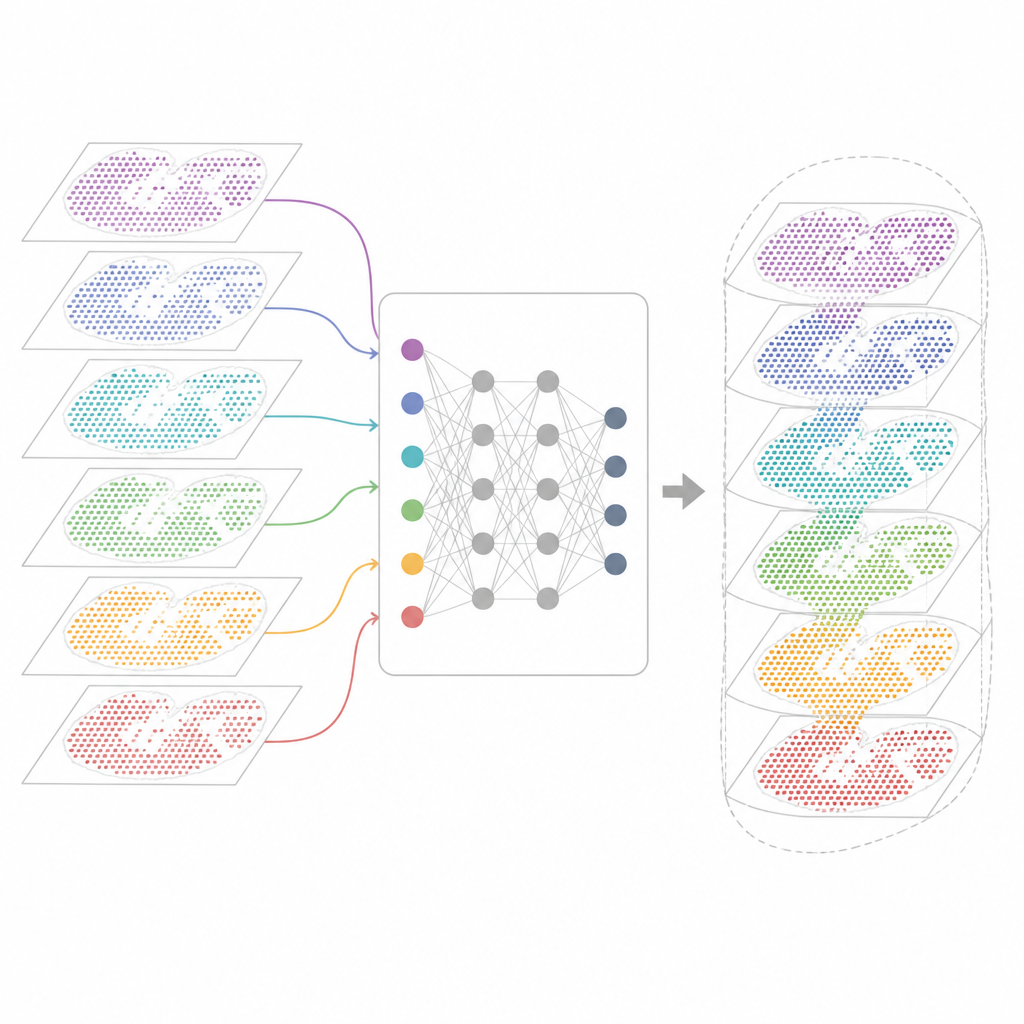
Por que Muitos Mapas Gênicos São Difíceis de Combinar
As tecnologias modernas de transcriptômica espacial medem a atividade gênica preservando a posição de cada célula no tecido. Alguns métodos capturam praticamente todos os genes, mas agregam várias células em cada ponto. Outros identificam células individuais, porém apenas para um painel selecionado de genes. Esses mapas também vêm de diferentes laboratórios, máquinas, momentos e espécies. Como resultado, cada conjunto de dados traz suas próprias peculiaridades técnicas e ruído. As ferramentas de análise existentes conseguem descrever uma lâmina ou algumas lâminas semelhantes, mas frequentemente falham quando se pede para alinhar dezenas de seções, transpor tecnologias ou acompanhar características teciduais tanto compartilhadas quanto específicas.
Uma Nova Estrutura para Unificar Mapas de Tecidos
O INSPIRE enfrenta esse problema de integração com uma estrutura de aprendizado profundo que respeita tanto as leituras gênicas quanto a disposição física das células. Primeiro ele constrói um “grafo de vizinhança” para cada lâmina de tecido, conectando pontos próximos. Uma rede neural baseada em grafos então transforma os dados brutos em uma representação interna compartilhada que mistura células comparáveis de diferentes seções, ao mesmo tempo em que permite que padrões específicos de cada seção permaneçam. Um componente adversarial atua como um crítico, identificando onde as seções ainda não se sobrepõem bem nesse espaço interno e impulsionando o modelo a melhorar o alinhamento.
De Padrões Ocultos a Atributos Legíveis por Humanos
Uma vez trazidos para esse espaço compartilhado, o INSPIRE decompõe o sinal remanescente em um conjunto de padrões espaciais recorrentes, chamados fatores, cada um ligado a um programa gênico característico. Isso é feito usando uma etapa de fatoração matricial não-negativa que incentiva o modelo a representar os dados como uma combinação de alguns blocos de construção simples. Cada fator corresponde a um padrão espacial através das lâminas, como uma camada específica do cérebro, um nicho tumoral ou uma região de órgão em desenvolvimento. Como o INSPIRE também aprende quais genes estão mais associados a cada fator, os pesquisadores podem interpretar esses padrões em termos de tipos celulares e processos biológicos conhecidos, em vez de números abstratos.
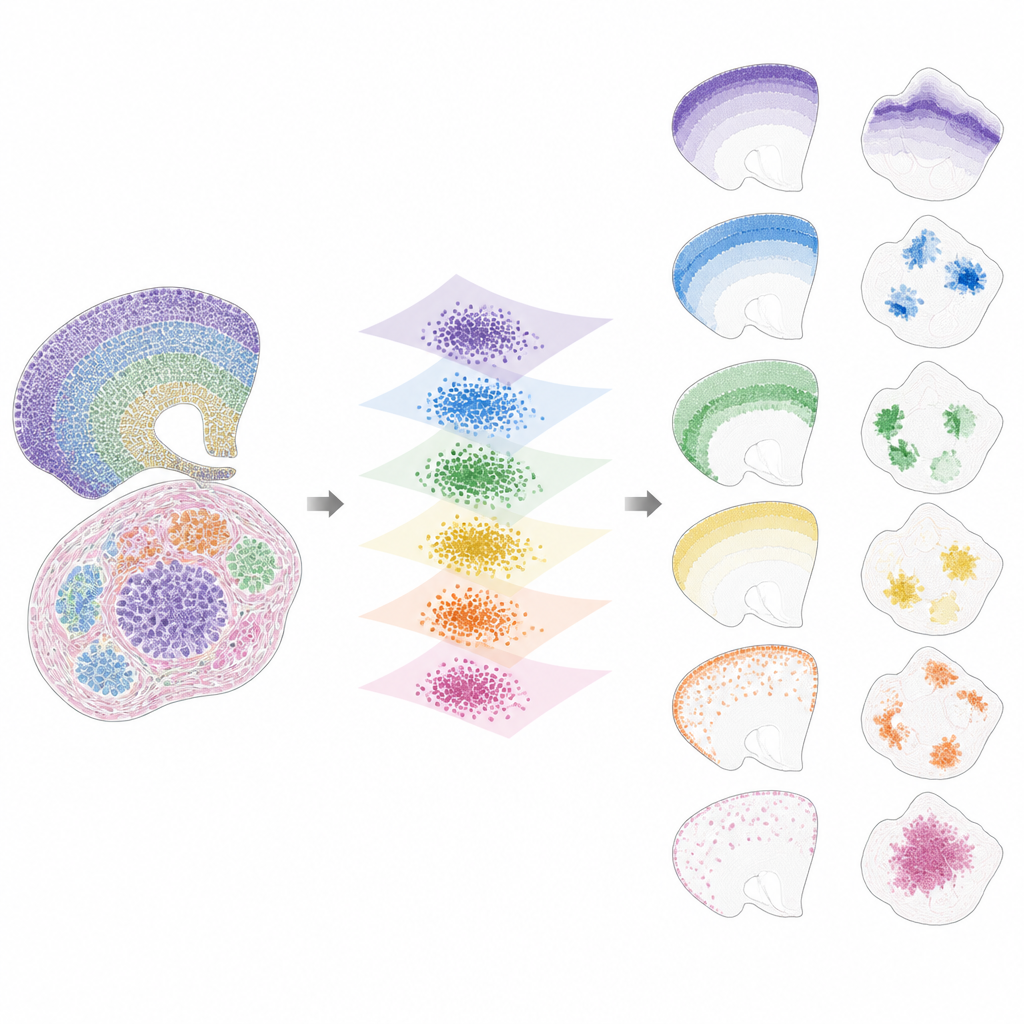
O que o INSPIRE Revela em Conjuntos de Dados Reais
Os autores mostram que o INSPIRE supera uma variedade de métodos populares tanto em conjuntos de dados simulados quanto reais. Em lâminas do córtex humano, ele alinha camadas entre doadores e recupera distinções finas entre tipos de neurônios e células de suporte, incluindo arranjos sutis que rótulos manuais perderam. No cérebro de camundongo, separa claramente estruturas compartilhadas, como o córtex, de regiões únicas como o cerebelo, identificando corretamente suas assinaturas gênicas distintas. O INSPIRE também faz a ponte entre diferentes tecnologias, combinando mapas de célula única com levantamentos mais amplos para transferir informações detalhadas de camadas e inferir padrões gênicos ausentes que nunca foram medidos diretamente.
Acompanhando Doença, Desenvolvimento e Estrutura 3D
Além de tecidos saudáveis, o INSPIRE expõe variação oculta em doença e desenvolvimento. Em seções humanas de câncer de mama com centenas de milhares de células, distingue regiões tumorais não invasivas de invasivas e revela subgrupos distintos de células de suporte ao redor de cada uma, ligados a marcadores conhecidos de agressividade e crescimento vascular. Em embriões de camundongo, o INSPIRE integra lâminas tomadas em múltiplos estágios para construir um atlas espaço-temporal, acompanhando como órgãos como coração, fígado, pulmão e cérebro crescem e se reorganizam. Ao alinhar com precisão lâminas adjacentes, também suporta reconstruções tridimensionais de órgãos e embriões inteiros, transformando pilhas de imagens bidimensionais em modelos coerentes de expressão gênica 3D.
O que Isso Significa para Estudos Futuros de Tecidos
Para um não especialista, o INSPIRE pode ser pensado como um tradutor poderoso que transforma muitos mapas gênicos imperfeitos e ruidosos em uma linguagem compartilhada de padrões espaciais e programas gênicos. Mantém o contexto de onde as células estão no tecido, filtra artefatos técnicos e destaca tanto características comuns quanto únicas entre experimentos. À medida que projetos de transcriptômica espacial ampliam para órgãos inteiros, tumores e organismos, métodos como o INSPIRE serão fundamentais para construir atlas integrados que os pesquisadores possam explorar para entender como as células cooperam, como as doenças perturbam a arquitetura tecidual e como estruturas complexas emergem durante o desenvolvimento.
Citação: Zhao, J., Zhang, X., Wang, G. et al. Interpretable, flexible and spatially aware integration of multiple spatial transcriptomics datasets from diverse sources. Nat Genet 58, 1138–1150 (2026). https://doi.org/10.1038/s41588-026-02579-x
Palavras-chave: transcriptômica espacial, arquitetura tecidual, mapas de expressão gênica, integração de dados, microambiente tumoral