Clear Sky Science · sv
YY1/Asprosin/PFKP‑axeln reglerar glykolytisk metabolism och förvärrar patologisk hjärtförtjockning
Varför hjärtats bränsleval spelar roll
Våra hjärtan förbrukar konstant bränsle för att hålla blodet i rörelse. Hos friska vuxna förlitar sig hjärtcellerna huvudsakligen på fett, med socker som en flexibel reserv. Hos många med långvarigt högt blodtryck eller annan belastning blir hjärtmuskeln tjockare och börjar till slut svikta. Denna studie ställer en enkel men viktig fråga för patienter och läkare: vad får ett stressat hjärta att ändra hur det använder bränsle, och kan vi upptäcka eller till och med avbryta det skiftet innan bestående skador uppstår?
En hormonsignal från vävnad till hjärta
Forskningen fokuserar på asprosin, ett hormon som frigörs från ett större protein kallat fibrillin och mest är känt för att hjälpa levern höja blodsockret under fasta. Teamet fann att personer med högt blodtryck och hjärtsvikt hade högre nivåer av asprosin i blodet än patienter utan svikt, och att dessa nivåer följde vanliga hjärtsviktsmarkörer som NT‑proBNP och ejektionsfraktion. Hos möss utsatta för trycköverbelastning, en vanlig modell för patologisk hjärtväxt, ökade asprosin i blodet men särskilt inne i hjärtat självt. Förvånande nog var det inte fettceller utan hjärtmuskelceller som blev den huvudsakliga källan till detta hormon under stress. 
När mer asprosin ger ett svagt och ärrat hjärta
För att testa orsakssamband använde forskarna genöverföringsverktyg för att antingen öka eller minska asprosin specifikt i musens hjärtmuskelceller. När de höjde asprosin och sedan tillförde trycköverbelastning, blev hjärtana större, pumpade mindre effektivt och utvecklade mer ärrvävnad och inflammation än kontrollhjärtan. Enskilda hjärtceller var tydligt förstorade och producerade mer klassiska markörproteiner för hjärtsvikt. Däremot skyddade nedreglering av asprosin genom att minska dess modergen FBN1 både manliga och kvinnliga möss från tryckinducerad förtjockning och stelhet. Dessa hjärtan behöll bättre pumpfunktion, hade mindre fibros och visade mindre cellstorlek, vilket tyder på att asprosin inte bara är en åskådare utan en aktiv drivkraft i skadlig ombyggnad.
Hur asprosin omprogrammerar sockernedbrytning i hjärtceller
Nästa steg var att förstå hur detta hormon ändrar cellernas beteende. Genaktivitet och metabola tester visade att asprosin skjuter hjärtceller mot snabbare sockernedbrytning, eller glykolys, samtidigt som deras mitokondrier — strukturerna som skapar det mesta av cellens ATP — försvagas. I odlade nyfödda mushjärtceller ökade tillförd asprosin eller artificiell överuttryckning syrabildning, glukosupptag och laktatutsläpp, alla tecken på upptrappad glykolys, samtidigt som totalt ATP minskade och mitokondriens membranpotential undergrävdes. Blockering av glykolys med en standardinhibitor utsuddade den celltillväxt som asprosin orsakar. I levande möss visade hjärtan utan asprosin under trycköverbelastning motsatt mönster, med mer balanserad bränsleanvändning och bättre energitillgång. 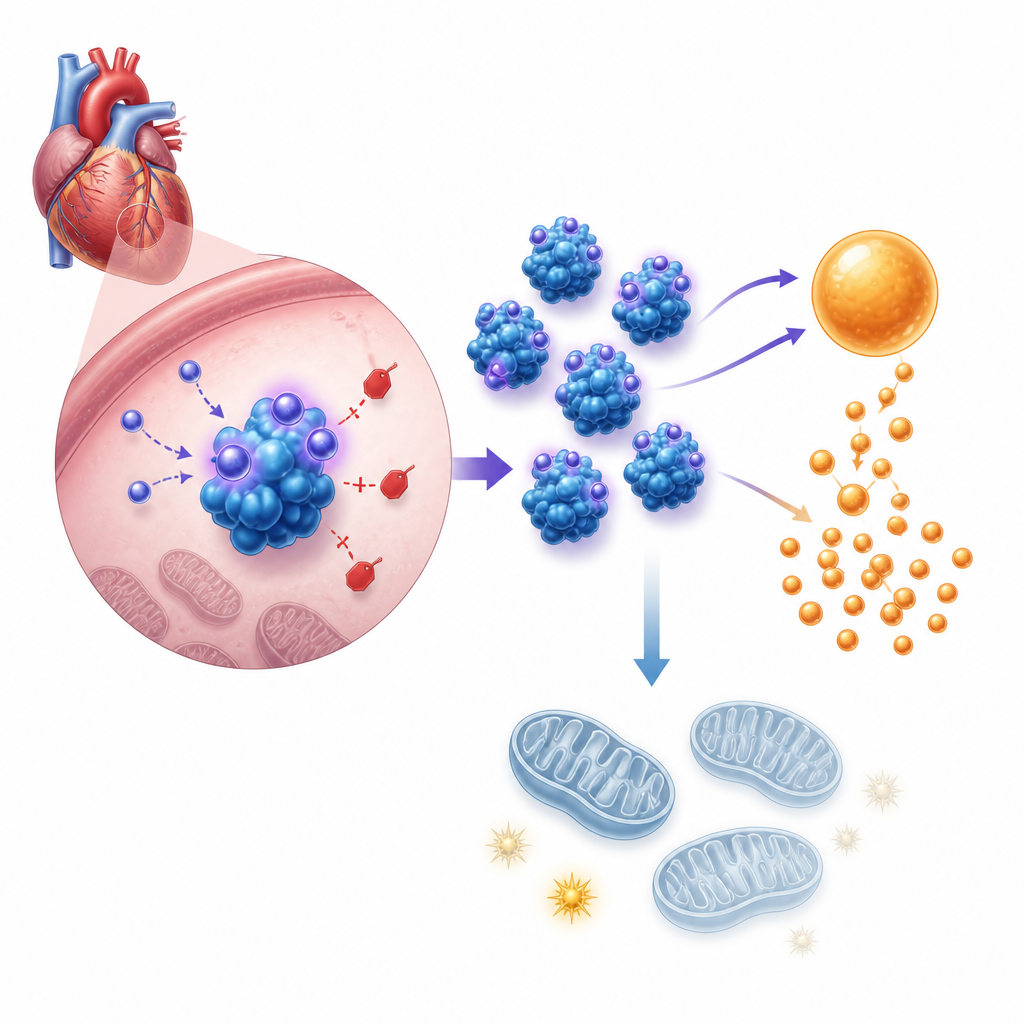
En proteinkedja som skyddar en viktig metabolisk omkopplare
Vidare upptäckte teamet att asprosin verkar genom att binda direkt till PFKP, ett huvudreglerande enzym i glykolysen. Under normala förhållanden märker ett enzym kallat DTX3L PFKP för destruktion via en process känd som K48‑länkad ubiquitinering, vilket håller dess nivåer i schack. Asprosin binder till PFKP på en specifik plats och hindrar DTX3L från att fästa dessa märken, så PFKP blir mer stabilt och mer rikligt. Detta ökade PFKP höjer i sin tur uttrycket av PDK4, vilket stänger av PDH‑enzymet som matar pyruvat in i mitokondrierna. Som ett resultat förbränns socker bara delvis till laktat, medan den fullständiga energiproducerande cykeln i mitokondrierna bromsas och ATP‑produktionen faller. Att ta bort PFKP i mus‑hjärtan lindrade tryckinducerad förstoring och fibros, och upphävde de skyddande effekterna av att sänka asprosin, vilket bevisar att PFKP är den avgörande mellanliggande länken.
Av/på‑knappen som sätter igång systemet
Slutligen undrade forskarna vad som slår på asprosin under hjärtstress. Genom DNA‑bindingsanalyser och biokemiska tester identifierade de transkriptionsfaktorn YY1 som en nyckelaktiverare av FBN1‑genen i hypertrofiska hjärtan. YY1‑nivåerna ökade i mus‑hjärtan efter trycköverbelastning och i hjärtceller behandlade med hormonet angiotensin II. YY1 band till en definierad plats i FBN1‑promotorn och ökade asprosinproduktionen. Att tysta YY1 dämpade förmågan hos extra asprosin att förstora hjärtceller och inducera sviktmarkörer, vilket placerar YY1 i toppen av en signalväg som löper genom asprosin, PFKP, PDK4 och PDH för att omforma energihanteringen.
Vad detta betyder för hjärtpatienter
Enkelt uttryckt beskriver detta arbete en bränsle‑kontrollaxel där hjärtstress ökar YY1, som slår på asprosin inne i hjärtcellerna. Asprosin skyddar därefter glykolysenzymet PFKP från nedbrytning, vilket tippar metabolismen mot snabb men ineffektiv sockerförbränning och bort från effektiv mitokondriell energiproduktion. Med tiden bidrar detta skifte till skadlig hjärtväxt och ärrbildning. Genom att identifiera asprosin i blodet som en möjlig tidig varningssignal och genom att kartlägga YY1–asprosin–PFKP–PDK4–PDH‑kedjan, pekar studien ut flera nya mål som en dag kan användas för att upptäcka eller mildra den metabola förskjutning som förvandlar ett hårt arbetande hjärta till ett sviktande.
Citering: Tong, M., Liu, X., Yu, Y. et al. YY1/Asprosin/PFKP axis regulates glycolytic metabolic and exacerbates pathological cardiac hypertrophy. Nat Commun 17, 4718 (2026). https://doi.org/10.1038/s41467-026-71197-2
Nyckelord: kardiell hypertrofi, hjärtats metabolism, asprosin, glykolys, mitokondriell energi