Clear Sky Science · nl
Het YY1/asprosin/PFKP‑as is reguleert glycolytische stofwisseling en verergert pathologische harthypertrofie
Waarom de brandstofkeuze van het hart telt
Onze harten verbranden voortdurend brandstof om het bloed in beweging te houden. Bij gezonde volwassenen gebruiken hartcellen voornamelijk vetten, met suiker als flexibele reserve. Bij veel mensen met langdurige hoge bloeddruk of andere belasting verdikt de hartspier en begint het hart tekort te schieten. Deze studie stelt een eenvoudige maar belangrijke vraag voor patiënten en artsen: wat veroorzaakt dat een gestrest hart zijn brandstofgebruik verandert, en kunnen we die verschuiving herkennen of zelfs onderbreken voordat er blijvende schade optreedt?
Een hormoonsignaal van vet naar hart
Het onderzoek richt zich op asprosin, een hormoon dat vrijkomt uit een groter eiwit genaamd fibrilline en vooral bekend is omdat het de lever helpt de bloedsuiker tijdens vasten te verhogen. Het team vond dat mensen met hoge bloeddruk en hartfalen hogere asprosinspiegels in hun bloed hadden dan patiënten zonder falen, en die niveaus correleerden met gangbare hartfalemarkers zoals NT‑proBNP en ejectiefractie. Bij muizen die onder drukoverbelasting werden geplaatst, een gangbaar model voor pathologische hartgroei, steeg asprosin in de bloedbaan maar vooral binnenin het hart zelf. Verrassend was dat het niet vetcellen waren maar hartspiercellen die onder stress de belangrijkste bron van dit hormoon werden. 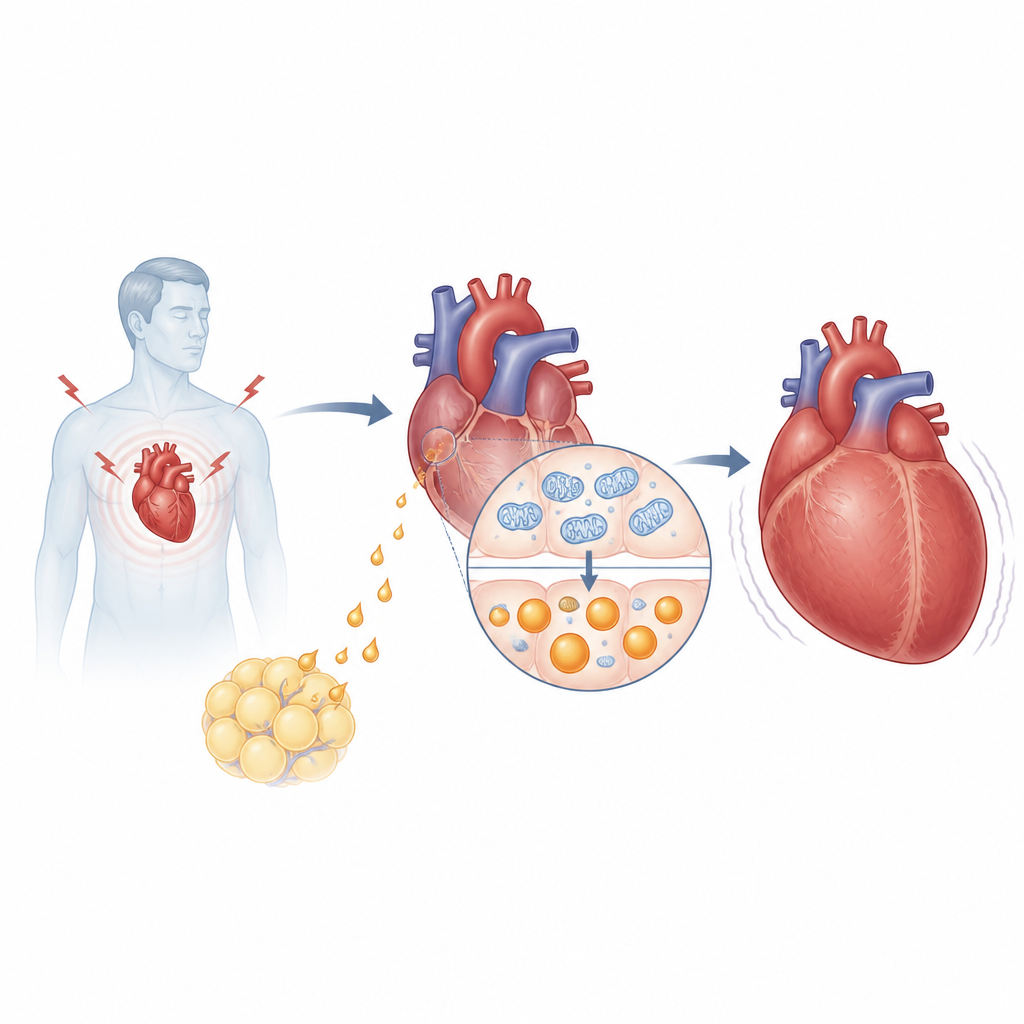
Meer asprosin maakt een zwak en littekentrijk hart
Om oorzaak en gevolg te testen, gebruikten de wetenschappers genleveringsmiddelen om asprosin specifiek in muishartspiercellen te verhogen of te verlagen. Wanneer ze asprosin ophoogden en vervolgens drukoverbelasting toepasten, werden de harten groter, pompten ze minder efficiënt en ontwikkelden ze meer littekenvorming en ontsteking dan controleharten. Individuele hartcellen waren duidelijk vergroot en produceerden meer klassieke hartfalemarkereiwitten. Daarentegen beschermde het terugschakelen van asprosin door vermindering van het oudergen FBN1 zowel mannelijke als vrouwelijke muizen tegen drukgeïnduceerde verdikking en verstijving. Deze harten hielden een betere pompfunctie, hadden minder littekenweefsel en toonden kleinere cellen, wat aangeeft dat asprosin niet slechts een bijstander is maar een actieve veroorzaker van schadelijke remodelering.
Hoe asprosin het suikerverbruik in hartcellen herschakelt
De volgende stap was begrijpen hoe dit hormoon het gedrag van cellen verandert. Genactiviteit en metabolische tests toonden aan dat asprosin hartcellen naar snellere suikerafbraak, of glycolyse, duwt, terwijl het tegelijkertijd hun mitochondriën verzwakt, de structuren die het grootste deel van het ATP‑energie van de cel produceren. In gekweekte pasgeboren muishartcellen verhoogde toegevoegde asprosin of kunstmatige overexpressie de zuurgraadproductie, glucoseopname en lactaatafgifte — allemaal tekenen van opgevoerde glycolyse — terwijl totale ATP afnam en de mitochondriale membraanpotentiaal werd ondermijnd. Het blokkeren van glycolyse met een standaardremmer maakte de celgroei veroorzaakt door asprosin ongedaan. In levende muizen vertoonden harten zonder asprosin onder drukoverbelasting het tegenovergestelde patroon, met evenwichtiger brandstofgebruik en een betere energietoevoer. 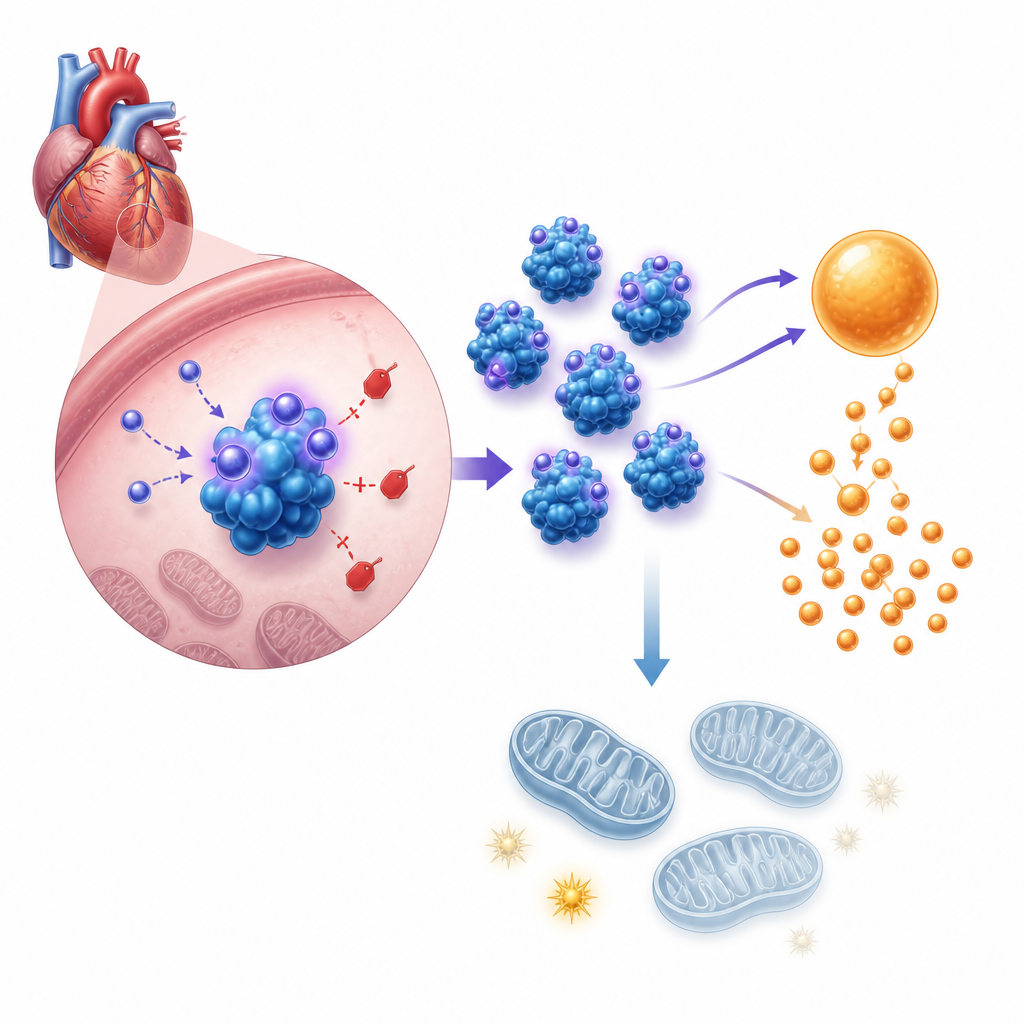
Een eiwitketen die een belangrijke metabole schakel bewaakt
Dieper gravend ontdekten de onderzoekers dat asprosin werkt door direct te binden aan PFKP, een belangrijk poorteiwit in de glycolyse. Onder normale omstandigheden labelt een enzym genaamd DTX3L PFKP voor afbraak via een proces dat K48‑gekoppelde ubiquitinatie wordt genoemd, waardoor de niveaus onder controle blijven. Asprosin bezet PFKP op een specifieke plaats en verhindert dat DTX3L deze labels aanbrengt, waardoor PFKP stabieler en talrijker wordt. Dit verhoogde PFKP doet vervolgens de expressie van PDK4 stijgen, dat het PDH‑enzym uitschakelt dat pyruvaat naar de mitochondriën voert. Daardoor wordt suiker slechts gedeeltelijk verbrand tot lactaat, terwijl de volledige energieproducerende cyclus in de mitochondriën vertraagt en de ATP‑output daalt. Het wegnemen van PFKP in muizharten verzachtte drukgeïnduceerde vergroting en fibrose, en het maakte de beschermende effecten van het verlagen van asprosin ongedaan, wat bewijst dat PFKP de cruciale schakel in het midden is.
De aan/uit‑schakelaar die het systeem activeert
Tenslotte vroegen de wetenschappers wat asprosin zelf inschakelt tijdens hartstress. Met DNAbindingsonderzoeken en biochemische tests identificeerden ze de transcriptiefactor YY1 als een belangrijke activator van het FBN1‑gen in hypertrofe harten. YY1‑niveaus stegen in muizharten na drukoverbelasting en in hartcellen behandeld met het hormoon angiotensine II. YY1 bond aan een bepaald gebied in de FBN1‑promotor en verhoogde de asprosinproductie. Het stilleggen van YY1 verzwakte het vermogen van extra asprosin om hartcellen te vergroten en faalmarkeringen te induceren, waarmee YY1 bovenaan een signaalketen staat die via asprosin, PFKP, PDK4 en PDH het energiemanagement hervormt.
Wat dit betekent voor hartpatiënten
In eenvoudige bewoordingen beschrijft dit werk een brandstofcontroledomein waarin hartstress YY1 verhoogt, wat asprosin in hartcellen activeert. Asprosin beschermt vervolgens het glycolyse‑enzym PFKP tegen afbraak, waardoor de stofwisseling kantelt naar snelle maar inefficiënte suikerverbranding en weg van efficiënte mitochondriale energieproductie. In de loop van de tijd draagt deze verschuiving bij aan schadelijke hartgroei en littekenvorming. Door asprosin in het bloed te identificeren als een mogelijke vroegtijdige waarschuwing en door de YY1–asprosin–PFKP–PDK4–PDH‑keten in kaart te brengen, wijst de studie op meerdere nieuwe doelen die mogelijk ooit kunnen worden gebruikt om de metabole neergang die een hardwerkend hart in een falend hart verandert, te detecteren of te verzachten.
Bronvermelding: Tong, M., Liu, X., Yu, Y. et al. YY1/Asprosin/PFKP axis regulates glycolytic metabolic and exacerbates pathological cardiac hypertrophy. Nat Commun 17, 4718 (2026). https://doi.org/10.1038/s41467-026-71197-2
Trefwoorden: cardiale hypertrofie, hartmetabolisme, asprosin, glycolyse, mitochondriale energie