Clear Sky Science · en
YY1/Asprosin/PFKP axis regulates glycolytic metabolic and exacerbates pathological cardiac hypertrophy
Why heart fuel choices matter
Our hearts are constantly burning fuel to keep blood moving. In healthy adults, heart cells rely mostly on fats, with sugar as a flexible backup. In many people with long term high blood pressure or other strain, the heart muscle thickens and begins to fail. This study asks a simple but important question for patients and doctors alike: what makes a stressed heart change the way it uses fuel, and can we spot or even interrupt that shift before lasting damage occurs?
A hormone signal from fat to the heart
The research focuses on asprosin, a hormone released from a larger protein called fibrillin and best known for helping the liver raise blood sugar during fasting. The team found that people with high blood pressure and heart failure had higher levels of asprosin in their blood than patients without failure, and those levels tracked with standard heart failure markers such as NT proBNP and ejection fraction. In mice subjected to pressure overload, a common model of pathological heart growth, asprosin rose in the bloodstream but especially inside the heart itself. Surprisingly, it was not fat cells but heart muscle cells that became the main source of this hormone under stress. 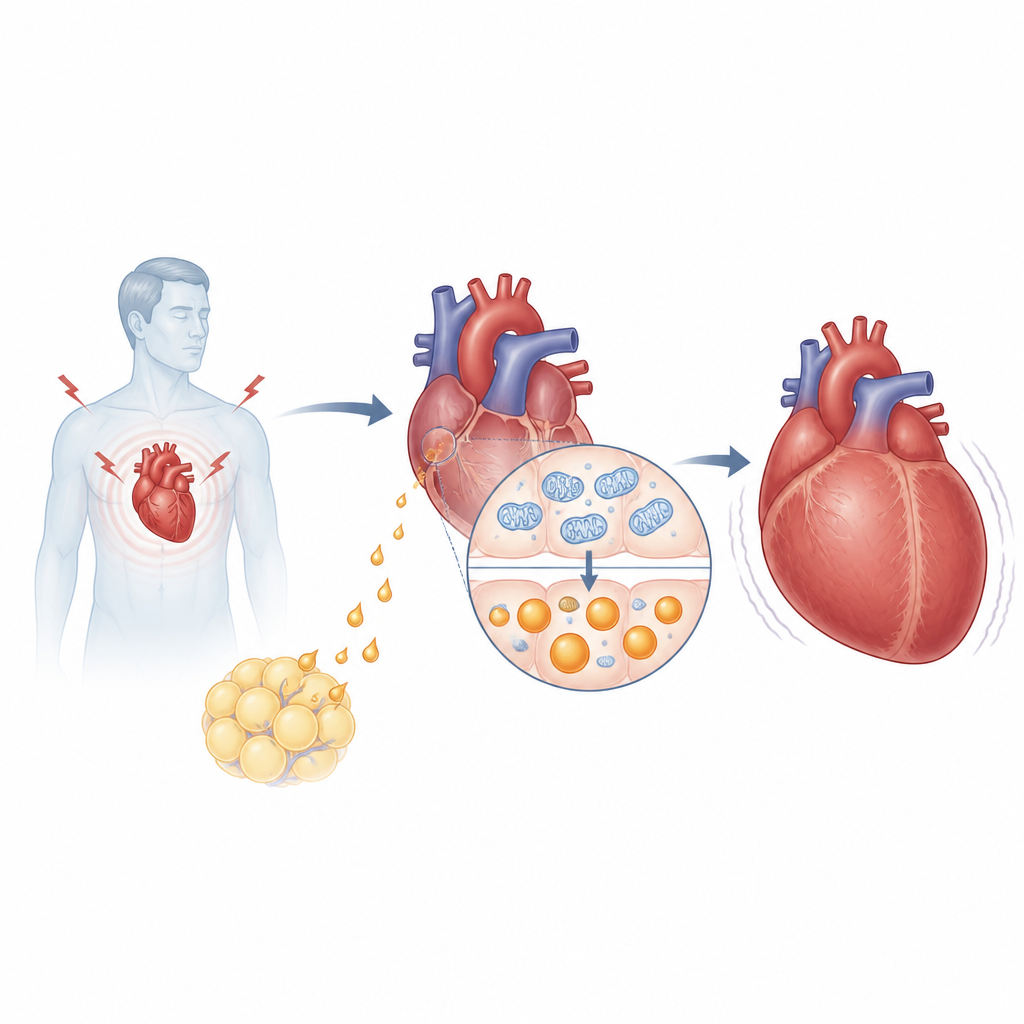
When more asprosin makes a weak and scarred heart
To test cause and effect, the scientists used gene delivery tools to either boost or reduce asprosin specifically in mouse heart muscle cells. When they drove up asprosin and then applied pressure overload, the hearts grew larger, pumped less efficiently, and developed more scarring and inflammation than control hearts. Individual heart cells were visibly enlarged and produced more classic heart failure marker proteins. In contrast, dialing down asprosin by reducing its parent gene FBN1 protected both male and female mice from pressure induced thickening and stiffening. These hearts kept better pumping function, had less scar tissue, and showed smaller cell size, indicating that asprosin is not just a bystander but an active driver of harmful remodeling.
How asprosin rewires sugar burning inside heart cells
The next step was to understand how this hormone changes cell behavior. Gene activity and metabolic tests showed that asprosin pushes heart cells toward faster sugar breakdown, or glycolysis, while at the same time weakening their mitochondria, the structures that make most of the cell’s ATP energy. In cultured newborn mouse heart cells, added asprosin or artificial overexpression increased acid production, glucose uptake, and lactate release, all signs of ramped up glycolysis, while lowering total ATP and undermining mitochondrial membrane potential. Blocking glycolysis with a standard inhibitor erased the cell growth caused by asprosin. In living mice, hearts lacking asprosin under pressure overload showed the opposite pattern, with more balanced fuel use and better energy supply. 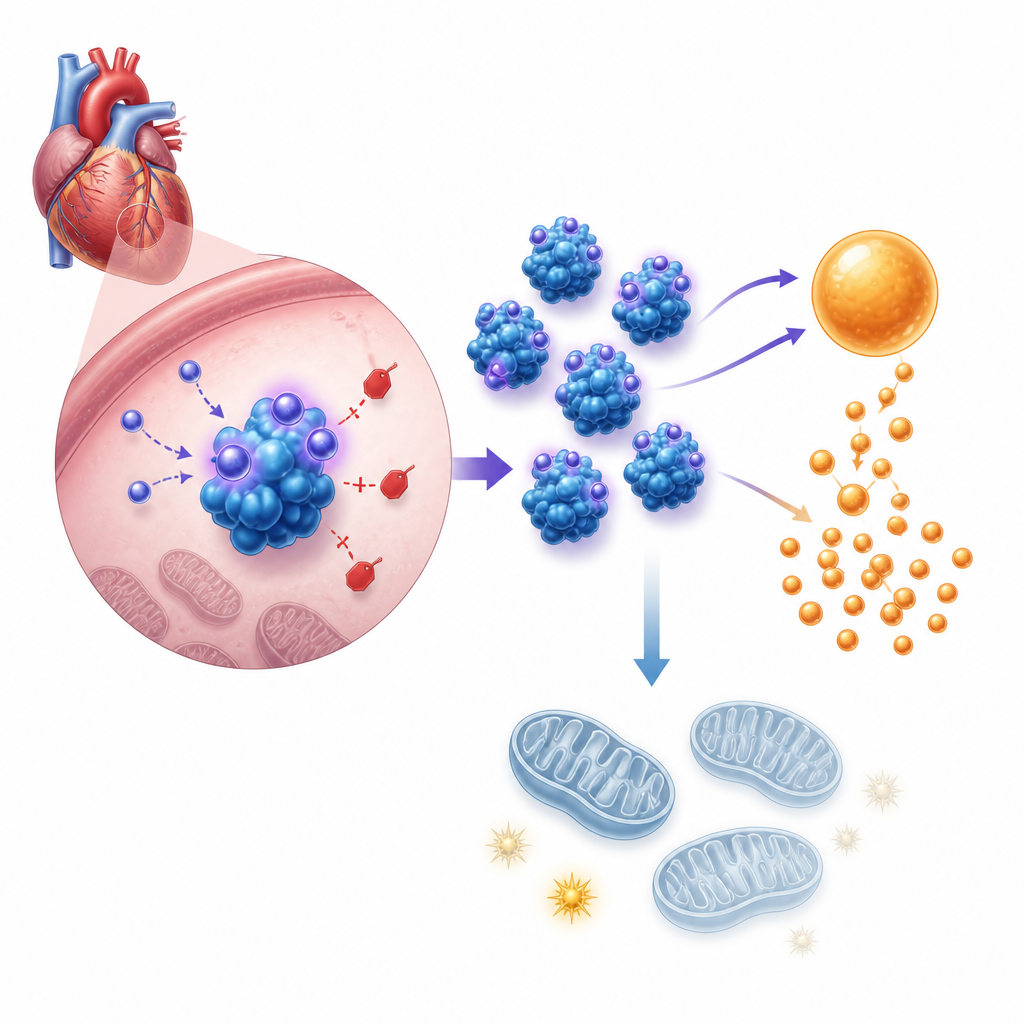
A protein chain that guards a key metabolic switch
Diving deeper, the team discovered that asprosin acts by binding directly to PFKP, a major gatekeeper enzyme in glycolysis. Under normal conditions, an enzyme called DTX3L tags PFKP for destruction through a process known as K48 linked ubiquitination, keeping its levels in check. Asprosin occupies PFKP at a specific site, preventing DTX3L from attaching these tags, so PFKP becomes more stable and abundant. This increased PFKP then raises expression of PDK4, which shuts down the PDH enzyme that feeds pyruvate into the mitochondria. As a result, sugar is burned only partway to lactate, while the full energy producing cycle in mitochondria slows and ATP output falls. Removing PFKP in mouse hearts eased pressure induced enlargement and fibrosis, and it cancelled out the protective effects of lowering asprosin, proving that PFKP is the crucial middle link.
The control switch that turns the system on
Finally, the scientists asked what flips asprosin itself on during heart stress. Using DNA binding surveys and biochemical tests, they identified the transcription factor YY1 as a key activator of the FBN1 gene in hypertrophic hearts. YY1 levels rose in mouse hearts after pressure overload and in heart cells treated with the hormone angiotensin II. YY1 bound a defined site in the FBN1 promoter and increased asprosin production. Silencing YY1 blunted the ability of extra asprosin to enlarge heart cells and induce failure markers, placing YY1 at the top of a signaling chain that runs through asprosin, PFKP, PDK4, and PDH to reshape energy handling.
What this means for heart patients
In plain terms, this work describes a fuel control axis in which heart stress boosts YY1, which turns on asprosin inside heart cells. Asprosin then shields the glycolysis enzyme PFKP from destruction, tipping metabolism toward quick but inefficient sugar burning and away from efficient mitochondrial energy production. Over time, this shift helps drive harmful heart growth and scarring. By identifying asprosin in the blood as a possible early warning sign and by outlining the YY1–asprosin–PFKP–PDK4–PDH chain, the study points to several new targets that might one day be used to detect or soften the metabolic slide that turns a hard working heart into a failing one.
Citation: Tong, M., Liu, X., Yu, Y. et al. YY1/Asprosin/PFKP axis regulates glycolytic metabolic and exacerbates pathological cardiac hypertrophy. Nat Commun 17, 4718 (2026). https://doi.org/10.1038/s41467-026-71197-2
Keywords: cardiac hypertrophy, heart metabolism, asprosin, glycolysis, mitochondrial energy