Clear Sky Science · de
YY1/Asprosin/PFKP-Achse reguliert glykolytischen Stoffwechsel und verschärft pathologische kardiale Hypertrophie
Warum die Brennstoffwahl des Herzens wichtig ist
Unser Herz verbrennt ständig Energie, um das Blut in Bewegung zu halten. Bei gesunden Erwachsenen nutzen Herzmuskelzellen überwiegend Fettsäuren, Zucker dient als flexible Reserve. Bei vielen Menschen mit dauerhaft hohem Blutdruck oder anderer Belastung verdickt sich der Herzmuskel und die Pumpleistung nimmt ab. Diese Studie stellt eine einfache, aber klinisch wichtige Frage: Was veranlasst ein gestresstes Herz, seine Brennstoffnutzung zu ändern, und lässt sich dieser Wechsel erkennen oder sogar unterbrechen, bevor bleibender Schaden entsteht?
Ein hormonelles Signal aus dem Fett- zum Herzgewebe
Im Fokus der Untersuchung steht Asprosin, ein Hormon, das aus dem größeren Protein Fibrillin freigesetzt wird und vor allem dafür bekannt ist, in Fastensituationen der Leber zu helfen, den Blutzucker anzuheben. Die Forscher fanden höhere Asprosinspiegel im Blut von Menschen mit Bluthochdruck und Herzinsuffizienz als bei Patienten ohne Herzversagen; diese Werte korrelierten mit etablierten Herzinsuffizienzmarkern wie NT-proBNP und der Ejektionsfraktion. In Mäusen, die einem Drucküberlastungs-Modell ausgesetzt wurden – ein gängiges Modell für pathologisches Herzwachstum –, stieg Asprosin im Blut, aber besonders stark im Herzen selbst an. Überraschenderweise waren es nicht Fettzellen, sondern die Herzmuskelzellen, die unter Stress zur hauptsächlichen Quelle dieses Hormons wurden. 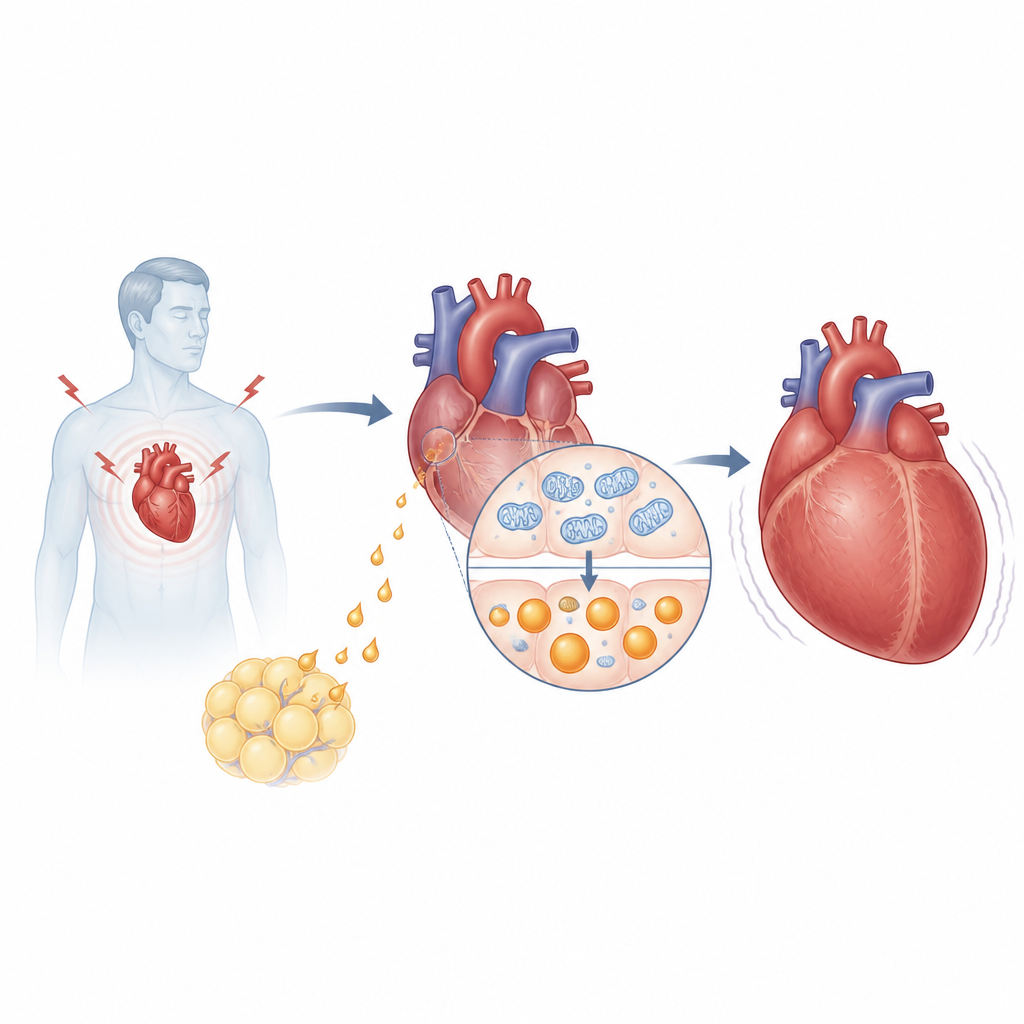
Mehr Asprosin führt zu einem schwächeren, vernarbten Herzen
Um Kausalität zu prüfen, verwendeten die Wissenschaftler gentherapeutische Werkzeuge, um Asprosin gezielt in den Herzmuskelzellen von Mäusen zu erhöhen oder zu reduzieren. Eine erhöhte Asprosinproduktion zusammen mit Drucküberlastung ließ die Herzen stärker wachsen, verschlechterte die Pumpfunktion und führte zu mehr Vernarbung und Entzündung als bei Kontrollen. Einzelne Herzmuskelzellen waren deutlich vergrößert und zeigten höhere Spiegel typischer Herzinsuffizienzmarker. Im Gegensatz dazu schützte das Herunterregeln von Asprosin durch Reduktion des Muttergens FBN1 sowohl männliche als auch weibliche Mäuse vor druckinduzierter Verdickung und Versteifung. Diese Herzen behielten eine bessere Pumpfunktion, wiesen weniger Narbengewebe auf und hatten kleinere Zellen, was darauf hinweist, dass Asprosin kein bloßer Begleiter, sondern ein aktiver Treiber schädlicher Umbauprozesse ist.
Wie Asprosin den Zuckerstoffwechsel in Herzmuskelzellen umstellt
Im nächsten Schritt untersuchten die Forscher, wie dieses Hormon das Zellverhalten verändert. Genexpressions- und Stoffwechseltests zeigten, dass Asprosin die Herzmuskelzellen in Richtung einer beschleunigten Zuckerzersetzung (Glykolyse) lenkt und gleichzeitig ihre Mitochondrien – die Hauptstätten der ATP-Produktion – schwächt. In kultivierten neugeborenen Maus-Herzmuskelzellen erhöhten zugefügtes Asprosin oder künstliche Überexpression die Säureproduktion, die Glukoseaufnahme und die Laktatfreisetzung – typische Zeichen einer gesteigerten Glykolyse – während die gesamte ATP-Menge sank und das mitochondriale Membranpotenzial beeinträchtigt wurde. Die Blockade der Glykolyse mit einem Standardinhibitor beseitigte das durch Asprosin ausgelöste Zellwachstum. In lebenden Mäusen zeigten Herzen ohne Asprosin bei Druckbelastung das gegenteilige Muster: ausgewogenere Brennstoffnutzung und bessere Energieversorgung. 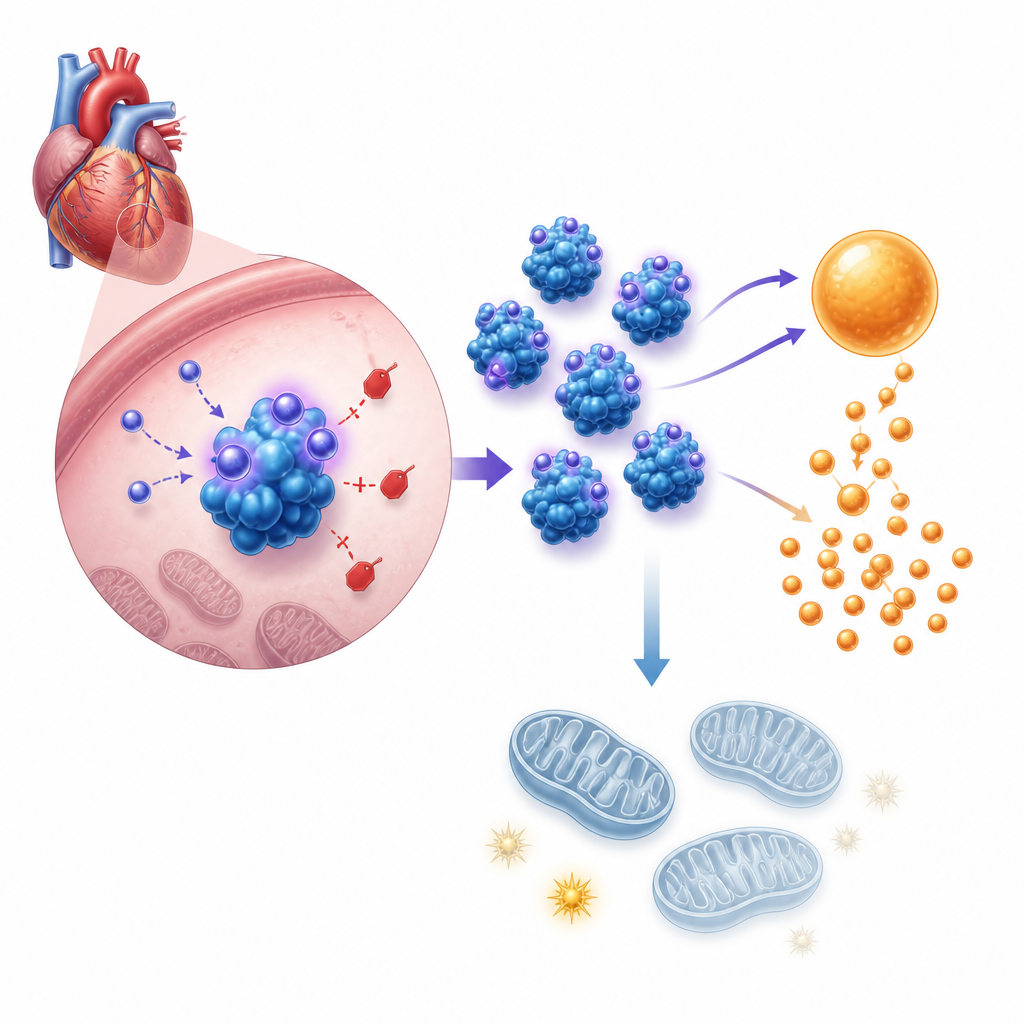
Eine Proteinkette, die einen zentralen Stoffwechselschalter schützt
Bei tiefergehenden Untersuchungen entdeckte das Team, dass Asprosin direkt an PFKP bindet, ein zentrales Glykalyse-Enzym und wichtiger Weichensteller. Unter normalen Bedingungen markiert ein Enzym namens DTX3L PFKP für den Abbau durch eine K48-verkettete Ubiquitinierung und hält dessen Menge so in Schach. Asprosin besetzt PFKP an einer spezifischen Stelle und verhindert damit, dass DTX3L diese Markierungen anbringt; folglich wird PFKP stabiler und häufiger. Das vermehrte PFKP steigert dann die Expression von PDK4, das das PDH-Enzym, welches Pyruvat in die Mitochondrien einspeist, hemmt. Dadurch wird Zucker nur teilweise zu Laktat verstoffwechselt, während der komplette energieerzeugende Kreislauf in den Mitochondrien verlangsamt und die ATP-Produktion reduziert wird. Das Entfernen von PFKP in Mausherzen milderte druckinduzierte Vergrößerung und Fibrose und hob die schützenden Effekte einer Asprosinreduktion auf – ein Beleg dafür, dass PFKP die entscheidende mittlere Verbindung ist.
Der Schaltknopf, der das System einschaltet
Schließlich fragten die Wissenschaftler, was Asprosin selbst bei Herzstress einschaltet. Mithilfe von DNA-Bindungsanalysen und biochemischen Tests identifizierten sie den Transkriptionsfaktor YY1 als wichtigen Aktivator des FBN1-Gens in hypertrophen Herzen. Die YY1-Spiegel stiegen in Mausherzen nach Drucküberlastung und in Herzmuskelzellen, die mit dem Hormon Angiotensin II behandelt wurden. YY1 band an eine definierte Stelle im FBN1-Promotor und erhöhte die Asprosinproduktion. Das Stilllegen von YY1 schwächte die Fähigkeit von zusätzlichem Asprosin, Herzmuskelzellen zu vergrößern und Versagensmarker zu induzieren, und positioniert YY1 damit an die Spitze einer Signalkette, die über Asprosin, PFKP, PDK4 und PDH die Energiehandhabung umgestaltet.
Was das für Herzpatienten bedeutet
Kurz gesagt beschreibt diese Arbeit eine Brennstoffsteuerungsachse, in der Herzstress YY1 hochreguliert und damit Asprosin in Herzmuskelzellen aktiviert. Asprosin schützt dann das Glykolyse-Enzym PFKP vor dem Abbau, verschiebt den Stoffwechsel hin zu schnellem, aber ineffizientem Zuckerverbrauch und weg von effizienter mitochondrialer Energieproduktion. Im Lauf der Zeit fördert diese Umstellung schädliches Herzwachstum und Vernarbung. Durch die Identifikation von Asprosin im Blut als möglichen Frühmarker und die Darstellung der YY1–Asprosin–PFKP–PDK4–PDH-Kette weist die Studie auf mehrere neue Ansatzpunkte hin, die eines Tages genutzt werden könnten, um das metabolische Abrutschen, das ein leistungsfähiges Herz in ein versagendes verwandelt, zu erkennen oder abzuschwächen.
Zitation: Tong, M., Liu, X., Yu, Y. et al. YY1/Asprosin/PFKP axis regulates glycolytic metabolic and exacerbates pathological cardiac hypertrophy. Nat Commun 17, 4718 (2026). https://doi.org/10.1038/s41467-026-71197-2
Schlüsselwörter: kardiale Hypertrophie, Herzstoffwechsel, Asprosin, Glykolyse, mitochondriale Energie