Clear Sky Science · it
L’asse YY1/Asprosin/PFKP regola il metabolismo glicolitico e aggrava l’ipertrofia cardiaca patologica
Perché le scelte di carburante del cuore sono importanti
I nostri cuori bruciano costantemente carburante per mantenere il flusso sanguigno. Negli adulti sani, le cellule cardiache si affidano prevalentemente ai grassi, con gli zuccheri come riserva flessibile. In molte persone con ipertensione prolungata o altri sforzi, il muscolo cardiaco si ispessisce e inizia a deteriorarsi. Questo studio pone una domanda semplice ma rilevante per pazienti e medici: cosa induce un cuore stressato a cambiare il modo in cui utilizza il carburante, e possiamo individuare o persino interrompere questo cambiamento prima che si verifichi un danno permanente?
Un segnale ormonale dal tessuto al cuore
La ricerca si concentra sull’asprosin, un ormone rilasciato da una proteina più grande chiamata fibrillina e noto soprattutto per il suo ruolo nell’aiutare il fegato ad aumentare la glicemia durante il digiuno. Il gruppo ha scoperto che le persone con ipertensione e insufficienza cardiaca presentavano livelli più alti di asprosin nel sangue rispetto ai pazienti senza insufficienza, e tali livelli correllavano con marcatori clinici standard dell’insufficienza cardiaca come NT-proBNP e frazione di eiezione. Nei topi sottoposti a sovraccarico di pressione, un modello comune di crescita cardiaca patologica, l’asprosin aumentava nel circolo sanguigno ma soprattutto all’interno del cuore stesso. Sorprendentemente, non sono state le cellule adipose ma le cellule del muscolo cardiaco a diventare la principale fonte di questo ormone sotto stress. 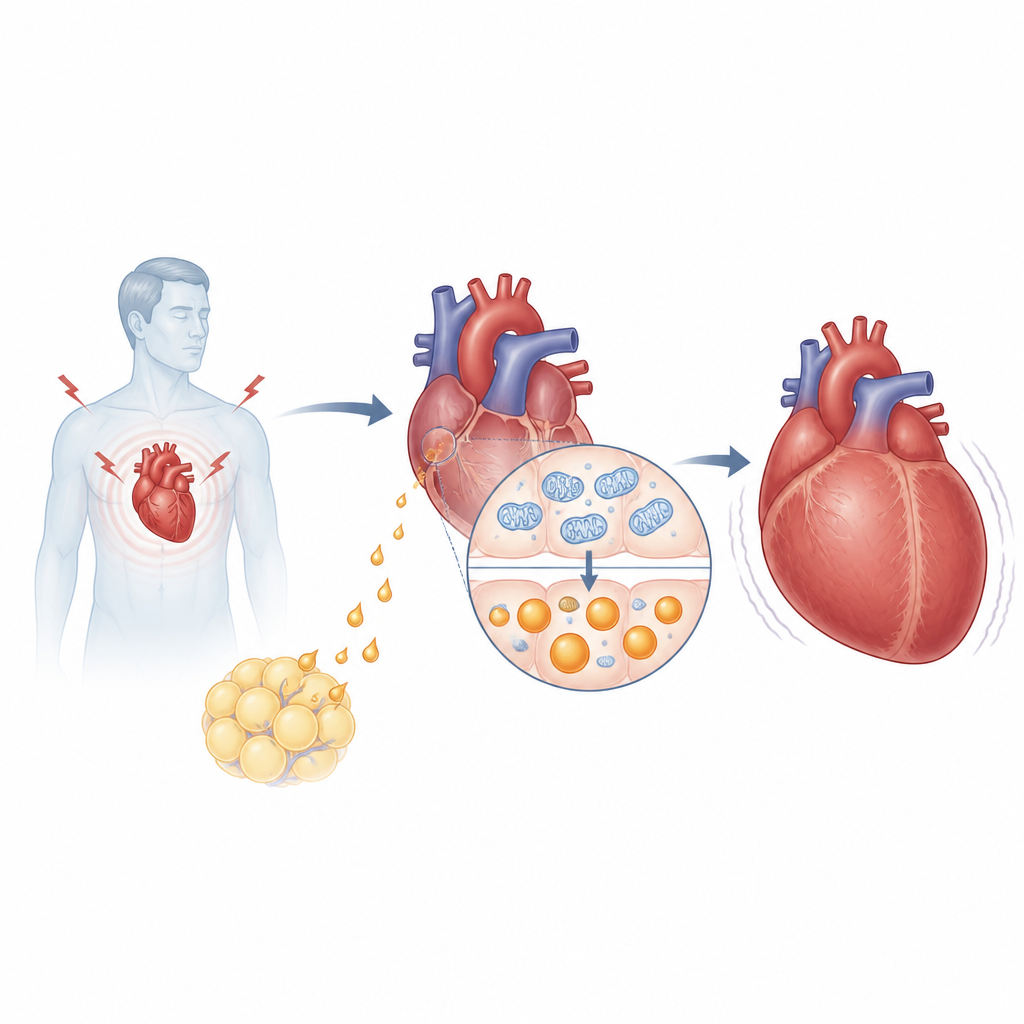
Quando più asprosin rende il cuore debole e fibrotico
Per testare nesso di causa ed effetto, gli scienziati hanno usato strumenti di consegna genica per aumentare o ridurre specificamente l’asprosin nelle cellule del muscolo cardiaco dei topi. Quando hanno incrementato l’asprosin e poi applicato il sovraccarico di pressione, i cuori sono cresciuti di volume, hanno pompato meno efficientemente e hanno sviluppato più fibrosi e infiammazione rispetto ai cuori di controllo. Le singole cellule cardiache apparivano ingrandite e mostravano una produzione aumentata di proteine tipiche dell’insufficienza cardiaca. Al contrario, abbassare l’asprosin riducendo il gene parentale FBN1 ha protetto sia i topi maschi sia femmine dall’ispessimento e dall’irrigidimento indotti dalla pressione. Questi cuori mantenevano una funzione di pompaggio migliore, avevano meno tessuto cicatriziale e dimensioni cellulari più contenute, indicando che l’asprosin non è un semplice spettatore ma un fattore attivo nella rimodellamento dannoso.
Come l’asprosin riorienta la combustione degli zuccheri nelle cellule cardiache
Il passo successivo è stato comprendere come questo ormone modifichi il comportamento cellulare. Analisi dell’espressione genica e test metabolici hanno mostrato che l’asprosin spinge le cellule cardiache verso una più rapida degradazione degli zuccheri, ovvero la glicolisi, indebolendo contemporaneamente i mitocondri, le strutture che producono la maggior parte dell’ATP cellulare. In cardiomiociti neonatali murini coltivati, l’aggiunta di asprosin o la sua sovraespressione artificiale aumentava la produzione di acidi, l’assorbimento di glucosio e il rilascio di lattato, tutti segnali di glicolisi potenziata, mentre diminuivano l’ATP totale e si comprometteva il potenziale di membrana mitocondriale. Bloccare la glicolisi con un inibitore standard annullava la crescita cellulare indotta dall’asprosin. Nei topi vivi, i cuori privi di asprosin sottoposti a sovraccarico di pressione mostravano il quadro opposto, con un uso del carburante più bilanciato e una migliore fornitura energetica. 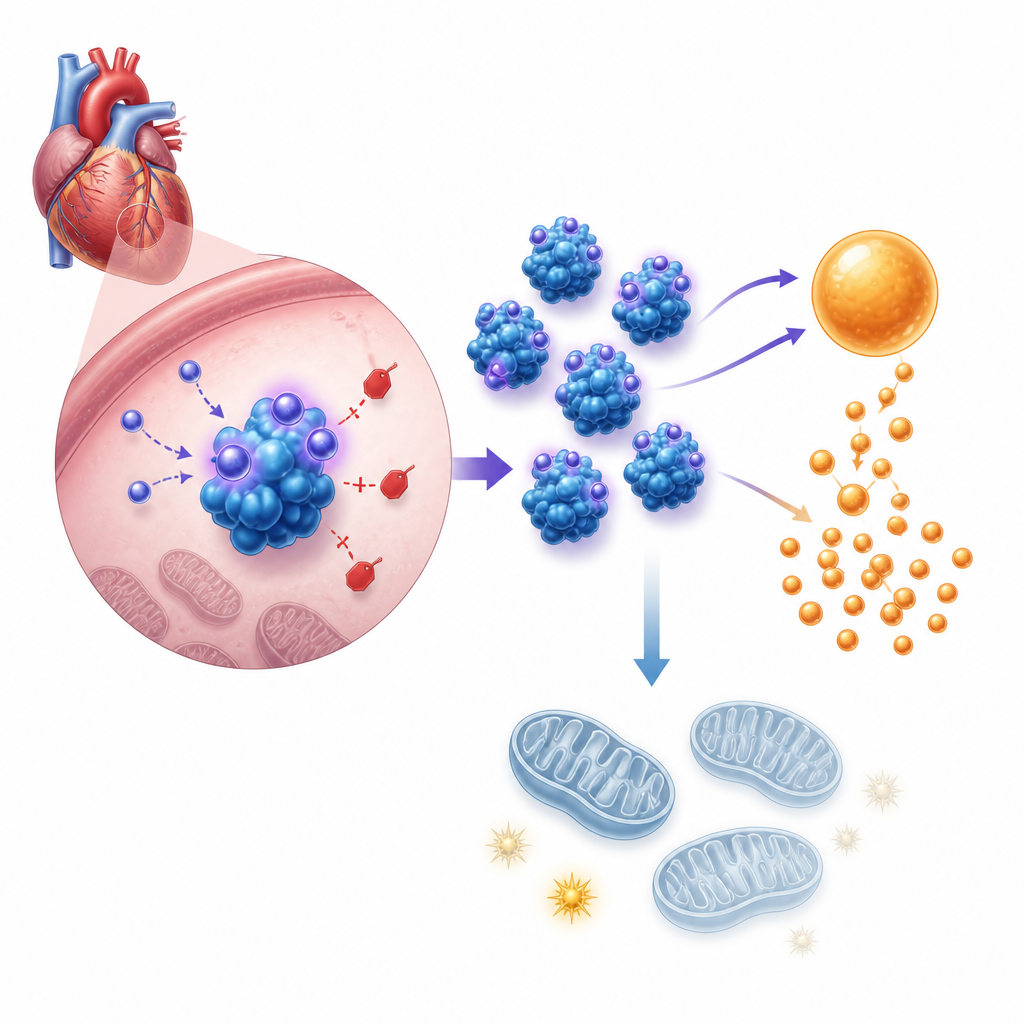
Una catena proteica che protegge un interruttore metabolico chiave
Scavando più a fondo, il team ha scoperto che l’asprosin agisce legandosi direttamente a PFKP, un importante enzima-guardiano nella glicolisi. In condizioni normali, un enzima chiamato DTX3L marca PFKP per la distruzione attraverso un processo noto come ubiquitinazione legata a K48, mantenendone i livelli sotto controllo. L’asprosin si lega a PFKP in un sito specifico, impedendo a DTX3L di attaccare questi marcatori, così PFKP diventa più stabile e abbondante. Questo aumento di PFKP poi alza l’espressione di PDK4, che spegne l’enzima PDH che convoglia il piruvato nei mitocondri. Di conseguenza, lo zucchero viene ossidato solo in parte fino al lattato, mentre il ciclo completo di produzione energetica mitocondriale rallenta e la produzione di ATP diminuisce. L’eliminazione di PFKP nei cuori dei topi attenuava l’ingrossamento e la fibrosi indotti dalla pressione, e cancellava gli effetti protettivi della riduzione di asprosin, dimostrando che PFKP è il collegamento cruciale centrale.
L’interruttore di controllo che accende il sistema
Infine, gli scienziati hanno cercato cosa attiva l’asprosin stesso durante lo stress cardiaco. Usando indagini di legame al DNA e test biochimici, hanno identificato il fattore di trascrizione YY1 come un attivatore chiave del gene FBN1 nei cuori ipertrofici. I livelli di YY1 aumentavano nei cuori murini dopo il sovraccarico di pressione e nelle cellule cardiache trattate con l’ormone angiotensina II. YY1 si legava a un sito definito nel promotore di FBN1 e incrementava la produzione di asprosin. L’azzeramento di YY1 attenuava la capacità dell’asprosin in eccesso di ingrandire le cellule cardiache e indurre marcatori di insufficienza, collocando YY1 in cima a una catena di segnalazione che passa attraverso asprosin, PFKP, PDK4 e PDH per rimodellare la gestione energetica.
Cosa significa per i pazienti cardiaci
In termini semplici, questo lavoro descrive un asse di controllo del carburante in cui lo stress cardiaco aumenta YY1, che attiva la produzione di asprosin all’interno delle cellule cardiache. L’asprosin poi protegge l’enzima della glicolisi PFKP dalla degradazione, spostando il metabolismo verso una combustione degli zuccheri rapida ma inefficiente e lontano dalla produzione energetica mitocondriale efficiente. Nel tempo, questo cambiamento contribuisce a guidare la crescita dannosa del cuore e la formazione di cicatrici. Identificando l’asprosin nel sangue come possibile segnale d’allarme precoce e delineando la catena YY1–asprosin–PFKP–PDK4–PDH, lo studio individua diversi nuovi bersagli che un giorno potrebbero essere utilizzati per rilevare o attenuare la deriva metabolica che trasforma un cuore laborioso in un cuore in insufficienza.
Citazione: Tong, M., Liu, X., Yu, Y. et al. YY1/Asprosin/PFKP axis regulates glycolytic metabolic and exacerbates pathological cardiac hypertrophy. Nat Commun 17, 4718 (2026). https://doi.org/10.1038/s41467-026-71197-2
Parole chiave: ipertrofia cardiaca, metabolismo cardiaco, asprosin, glicolisi, energia mitocondriale