Clear Sky Science · pl
Wzlot i upadek inhibitorów wymiany zasadowej SARM1
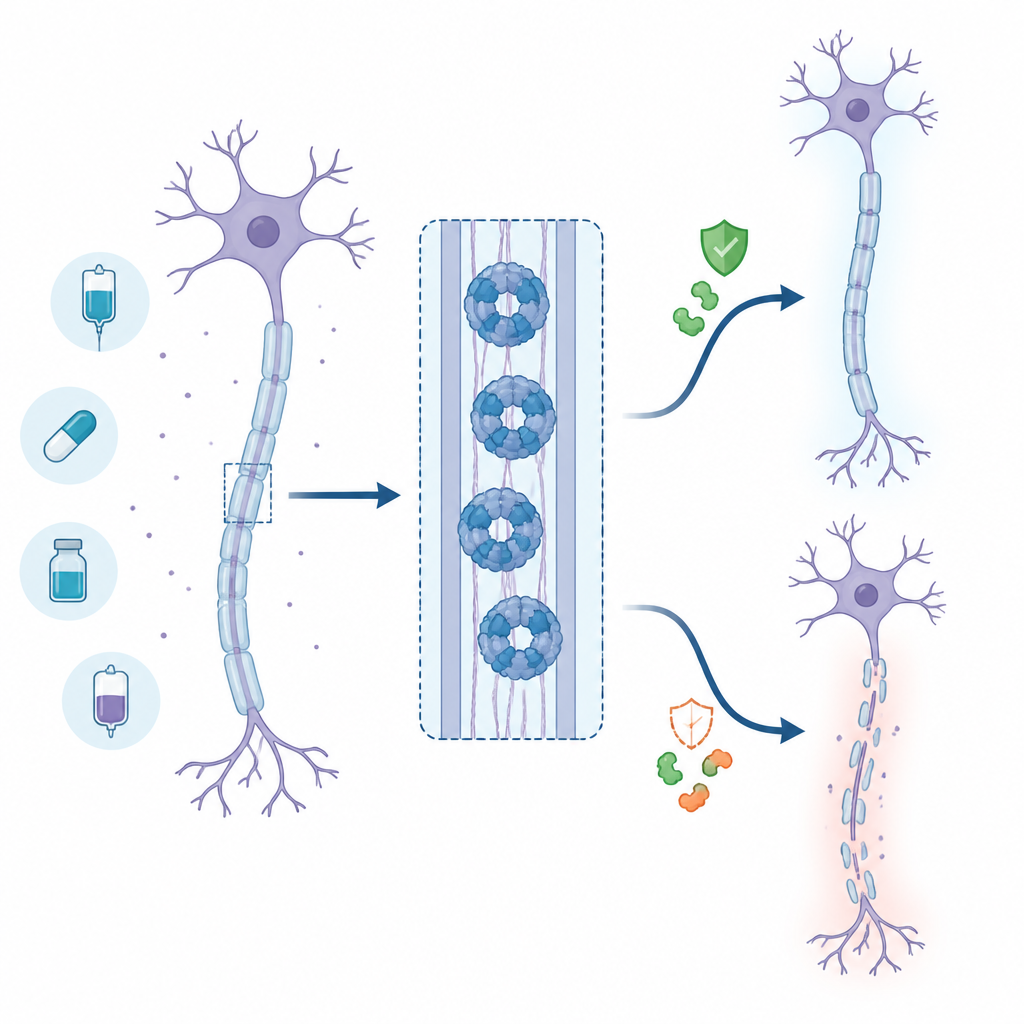
Dlaczego ochrona nerwów ma znaczenie
Wielu pacjentów onkologicznych i osób z chorobami neurologicznymi cierpi z powodu uszkodzeń długich „przewodów” komórek nerwowych, zwanych aksonami. Uszkodzenia te mogą powodować ból, drętwienie lub słabość, które utrzymują się przez lata. Naukowcy z dużym zainteresowaniem przyglądają się białku nazwanemu SARM1, które wydaje się działać jak przełącznik samodestrukcji aksonów w warunkach stresu. Gdyby udało się bezpiecznie wyłączyć SARM1 za pomocą leków, można by spowolnić lub zapobiec uszkodzeniom nerwów. To badanie śledzi jedną z takich strategii lekowych i wyjaśnia, dlaczego początkowo obiecujące podejście ostatecznie musiało zostać porzucone.
Przełącznik samodestrukcji nerwu
Aksony polegają na stałym dopływie energii, napędzanym przez cząsteczkę zwaną NAD+. Kiedy akson zostaje uszkodzony lub zestresowany, mechanizmy zaopatrujące go w NAD+ zawodzą i poziom NAD+ spada. SARM1 wykrywa ten spadek i po aktywacji szybko rozkłada pozostały NAD+, wpędzając akson w kryzys energetyczny i strukturalny. Badania na zwierzętach, w których SARM1 został usunięty, pokazują silną ochronę nerwów po urazie lub chemioterapii, co uczyniło SARM1 priorytetowym celem w poszukiwaniach leków.
Polowanie na nową klasę blokerów
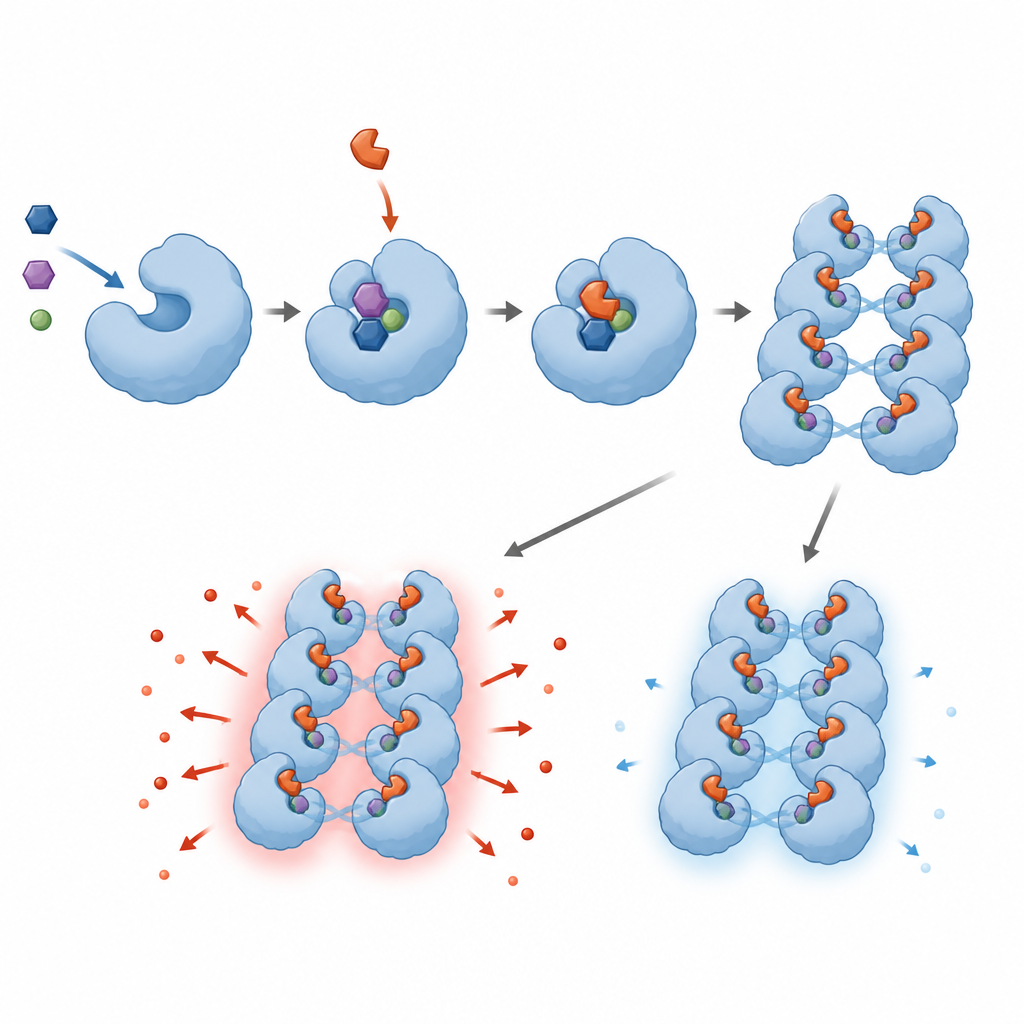
Naukowcy przesiewali 1,8 miliona małych cząsteczek, aby znaleźć te, które potrafią zablokować ludzki SARM1 w probówce. Odkryli rodzinę związków nazywanych inhibitorami wymiany zasadowej, które wchodzą w interakcję bezpośrednio z miejscem aktywnym SARM1 i są przez enzym przekształcane w addukty przypominające NAD+, które pozostają w nim uwięzione. Te addukty wyłączają zdolność SARM1 do rozkładu NAD+ w testach biochemicznych i chroniły komórki przypominające neurony oraz neuronsensoryczne komórki gryzoni przed uszkodzeniem wywołanym lekami chemioterapeutycznymi lub neurotoksyną. Zespół udoskonalił struktury chemiczne, by poprawić siłę działania, rozpuszczalność i bezpieczeństwo, oraz potwierdził dopasowanie związków do SARM1 za pomocą zaawansowanych narzędzi, takich jak kriomikroskopia elektronowa i wymiana wodór–deuter.
Ukryty zwrot akcji przy niskich poziomach leku
Poza standardowymi warunkami przesiewu zespół odkrył nieoczekiwane i niepokojące zachowanie. Gdy inhibitory wymiany zasadowej stosowano w dawkach zbyt niskich, by całkowicie zablokować SARM1, nie stawały się po prostu nieskuteczne — w rzeczywistości zwiększały aktywność SARM1. W hodowlach neuronów pochodzenia gryzoni i ludzi takie podinhibicyjne poziomy powodowały szybszą degenerację aksonów, co odpowiadało wyższym poziomom cząsteczki sygnałowej generowanej przez SARM1. W zmodyfikowanych liniach komórkowych niskie dawki tych samych leków przyspieszały utratę NAD+ zamiast jej zapobiegać. Szczegółowe badania strukturalne i biofizyczne sugerują, że addukty pochodzące od inhibitora sprzyjają skupianiu się jednostek SARM1 w rozległe zespoły, zwiększając ogólną aktywność enzymu, chyba że niemal wszystkie miejsca są zajęte.
Co się dzieje u żywych zwierząt
Aby sprawdzić, czy ten paradoks występuje w organizmie, badacze przetestowali zaawansowany inhibitor wymiany zasadowej, już używany przez inne grupy, w modelu mysim urazu nerwu kulszowego. Na początku leczone myszy wykazywały niższe poziomy krążącego markera uszkodzenia aksonów, co sugerowało tymczasową ochronę przy wysokich stężeniach leku. Jednak w miarę jak związek był usuwany z organizmu, poziomy markera odbiły się i nawet przewyższyły te u zwierząt nieleczonych, co jest zgodne z opóźnionym, lecz ostatecznie silniejszym uszkodzeniem nerwów. W połączeniu z podobnymi doniesieniami z innych laboratoriów, wyniki te wskazują na ryzyko w świecie rzeczywistym, że częściowe dawkowanie tej klasy związków mogłoby pogorszyć zamiast złagodzić uraz nerwu.
Co to oznacza dla przyszłych leków ratujących nerwy
Praca ta pokazuje, że choć SARM1 pozostaje atrakcyjnym celem zapobiegania utracie aksonów, inhibitory wymiany zasadowej prawdopodobnie nie będą bezpiecznymi lekami. Ponieważ są one wytwarzane i wiązane przez samego SARM1, mogą działać jak molekularny klej, który najpierw wycisza, a potem nadaktywuje białko, które mają kontrolować, chyba że poziomy leku są utrzymywane stale na wysokim poziomie. Autorzy w związku z tym wstrzymali rozwój tej rodziny związków i argumentują, że przyszłe wysiłki powinny skupić się na innych sposobach modulacji SARM1, takich jak celowanie w jego domeny regulacyjne lub wykorzystanie podejść genetycznych. Ich doświadczenie podkreśla, jak ważne jest łączenie potężnych narzędzi przesiewowych z dokładnymi badaniami mechanistycznymi, zanim leki chroniące nerwy trafią do kliniki.
Cytowanie: Lundbäck, T., Chandrasekar, V., Gu, C. et al. The rise and fall of SARM1 base-exchange inhibitors. Commun Chem 9, 183 (2026). https://doi.org/10.1038/s42004-026-02074-8
Słowa kluczowe: SARM1, degeneracja aksonów, neuropatia wywołana chemioterapią, neuroprotekcja, metabolizm NAD