Clear Sky Science · en
The rise and fall of SARM1 base-exchange inhibitors
Why nerve protection matters
Many cancer patients and people with neurological diseases suffer from damage to the long wires of nerve cells, called axons. This damage can cause pain, numbness, or weakness that may last for years. Scientists have been excited about a protein named SARM1, which appears to act as a self‑destruct switch for axons under stress. If drugs could safely turn SARM1 off, they might slow or prevent nerve damage. This study follows one such drug approach, explaining why an initially promising strategy ultimately had to be abandoned.
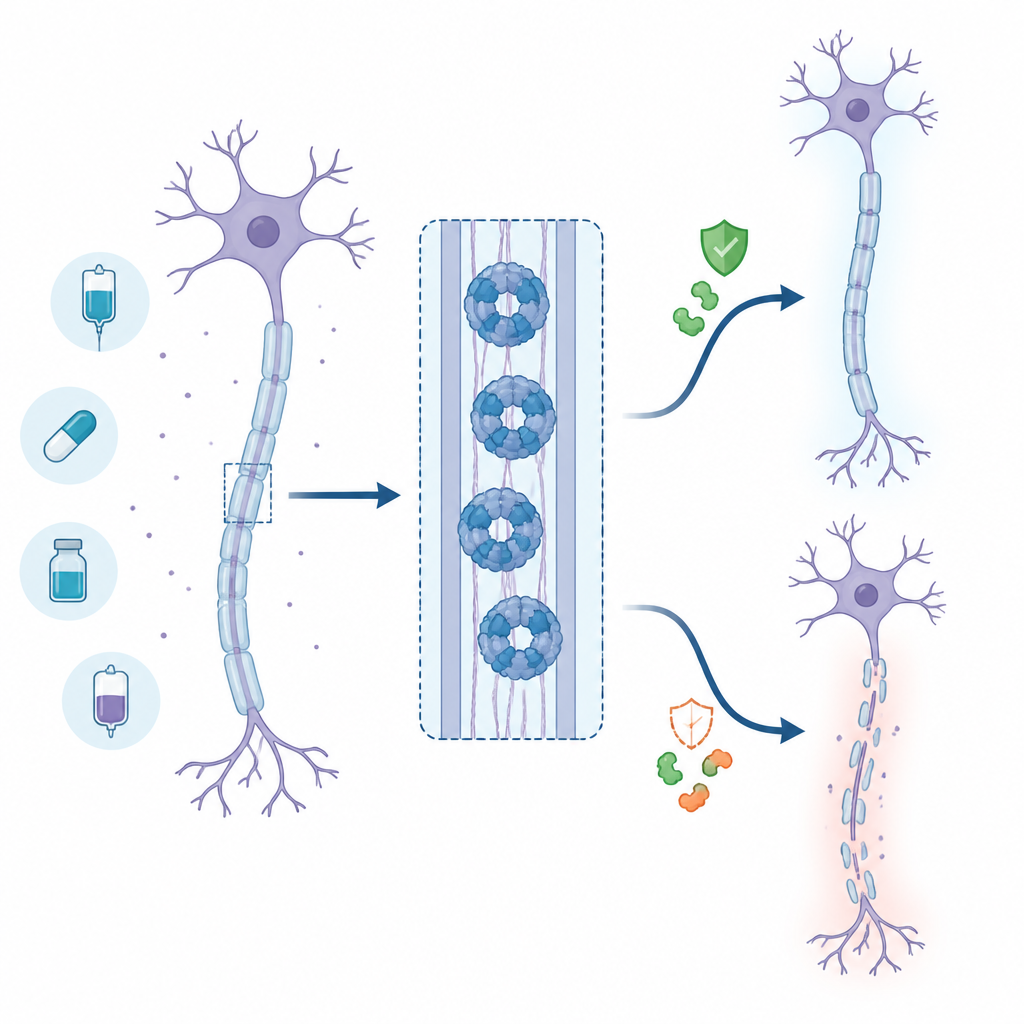
The nerve self‑destruct switch
Axons depend on a steady energy supply, fuelled by a molecule called NAD+. When an axon is injured or stressed, its supply machinery breaks down and NAD+ levels fall. SARM1 senses this drop and, once activated, rapidly chews up remaining NAD+, pushing the axon into an energy crisis and structural breakdown. Animal studies where SARM1 is removed show strong protection of nerves after injury or chemotherapy, which made SARM1 a high‑priority target for drug discovery.
Hunting for a new class of blockers
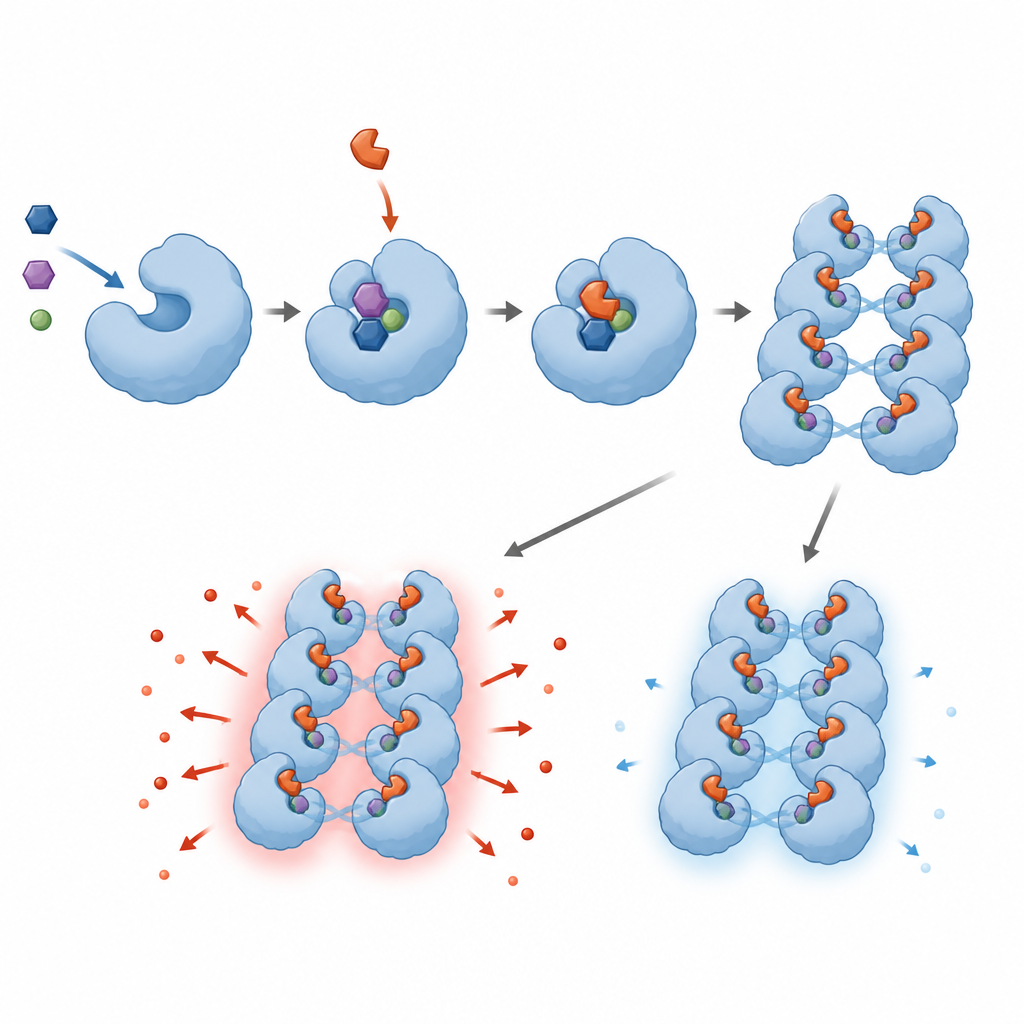
The researchers screened 1.8 million small molecules to find those that could block human SARM1 in the test tube. They discovered a family of compounds, called base‑exchange inhibitors, that interact directly with SARM1’s active site and are turned by the enzyme into NAD‑like adducts that stay lodged inside it. These adducts shut down SARM1’s ability to break down NAD+ in biochemical tests and protected nerve‑like cells and rodent sensory neurons from damage caused by chemotherapy drugs or a neurotoxin. The team refined the chemical structures to improve potency, solubility, and safety, and confirmed how the compounds fit into SARM1 using advanced tools such as cryo‑electron microscopy and hydrogen–deuterium exchange.
Hidden twist at low drug levels
As the team pushed beyond standard screening conditions, they uncovered an unexpected and troubling behavior. When base‑exchange inhibitors were used at doses too low to fully block SARM1, they did not simply become ineffective; they actually made SARM1 more active. In rodent and human neuron cultures, these sub‑inhibitory levels caused axons to degenerate faster, matched by higher levels of a SARM1‑generated signal molecule. In engineered cell lines, low doses of the same drugs sped up the loss of NAD+ instead of preventing it. Detailed structural and biophysical work suggests that the inhibitor‑derived adducts encourage SARM1 units to cluster into extended assemblies, increasing overall enzyme activity unless almost every site is occupied.
What happens in living animals
To see whether this paradox appears in the body, the researchers tested an advanced base‑exchange inhibitor, already used by other groups, in a mouse model of sciatic nerve injury. Early on, the treated mice showed lower levels of a blood marker of axon damage, suggesting temporary protection while drug levels were high. However, as the compound cleared from the body, marker levels rebounded and even rose above those in untreated animals, consistent with delayed but ultimately stronger nerve damage. Combined with similar reports from other laboratories, these results point to a real‑world risk that partial dosing of this drug class could worsen rather than ease nerve injury.
What this means for future nerve‑saving drugs
This work shows that while SARM1 remains an attractive target for preventing axon loss, base‑exchange inhibitors are unlikely to be safe medicines. Because they are made and bound by SARM1 itself, they can act like a molecular glue that first silences and then over‑activates the very protein they are meant to control, unless drug levels are kept continuously high. The authors therefore halted development of this compound family and argue that future efforts should focus on different ways to modulate SARM1, such as targeting its regulatory domains or using genetic approaches. Their experience highlights how important it is to pair powerful screening tools with careful mechanistic studies before moving nerve‑protective drugs into the clinic.
Citation: Lundbäck, T., Chandrasekar, V., Gu, C. et al. The rise and fall of SARM1 base-exchange inhibitors. Commun Chem 9, 183 (2026). https://doi.org/10.1038/s42004-026-02074-8
Keywords: SARM1, axon degeneration, chemotherapy-induced neuropathy, neuroprotection, NAD metabolism