Clear Sky Science · de
Aufstieg und Fall der SARM1-Base‑Exchange‑Inhibitoren
Warum Nervenschutz wichtig ist
Viele Krebspatienten und Menschen mit neurologischen Erkrankungen leiden unter Schäden an den langen Leitungen von Nervenzellen, den Axonen. Diese Schäden können Schmerzen, Taubheit oder Schwäche hervorrufen, die Jahre anhalten können. Forschende sind von einem Protein namens SARM1 begeistert, das offenbar als Selbstzerstörungsschalter für Axone unter Stress fungiert. Wenn Medikamente SARM1 sicher abschalten könnten, ließe sich Nervenschaden möglicherweise verlangsamen oder verhindern. Diese Studie verfolgt einen solchen Therapieansatz und erklärt, warum eine zunächst vielversprechende Strategie letztlich aufgegeben werden musste.
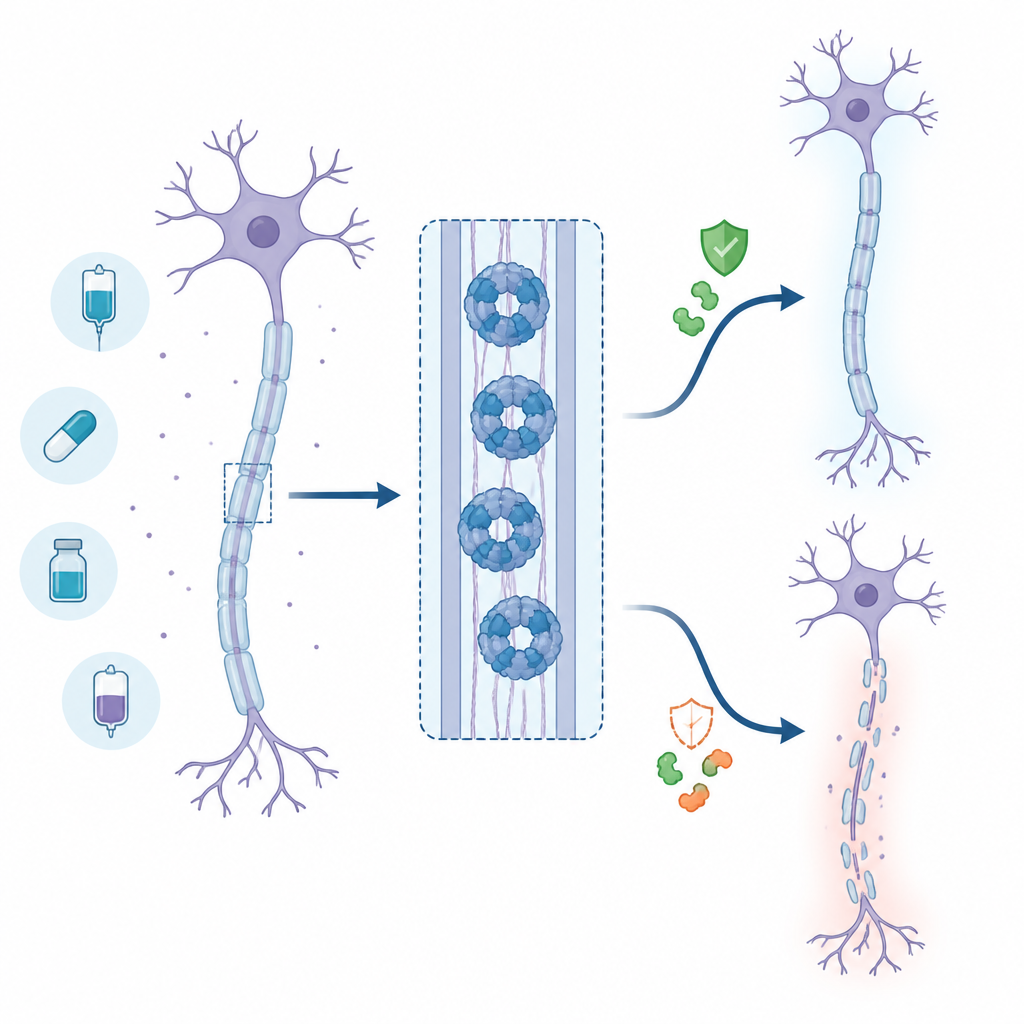
Der neurale Selbstzerstörungsschalter
Axone sind auf eine stetige Energiezufuhr angewiesen, die durch ein Molekül namens NAD+ bereitgestellt wird. Wenn ein Axon verletzt oder gestresst ist, versagt seine Versorgungsmaschinerie und die NAD+-Spiegel sinken. SARM1 erkennt diesen Abfall und baut, einmal aktiviert, rasch das verbleibende NAD+ ab, was das Axon in eine Energiekrise und strukturellen Zerfall treibt. Tierversuche, in denen SARM1 entfernt wird, zeigen starken Schutz der Nerven nach Verletzung oder Chemotherapie, weshalb SARM1 ein vorrangiges Ziel für die Wirkstoffsuche wurde.
Auf der Suche nach einer neuen Klasse von Blockern
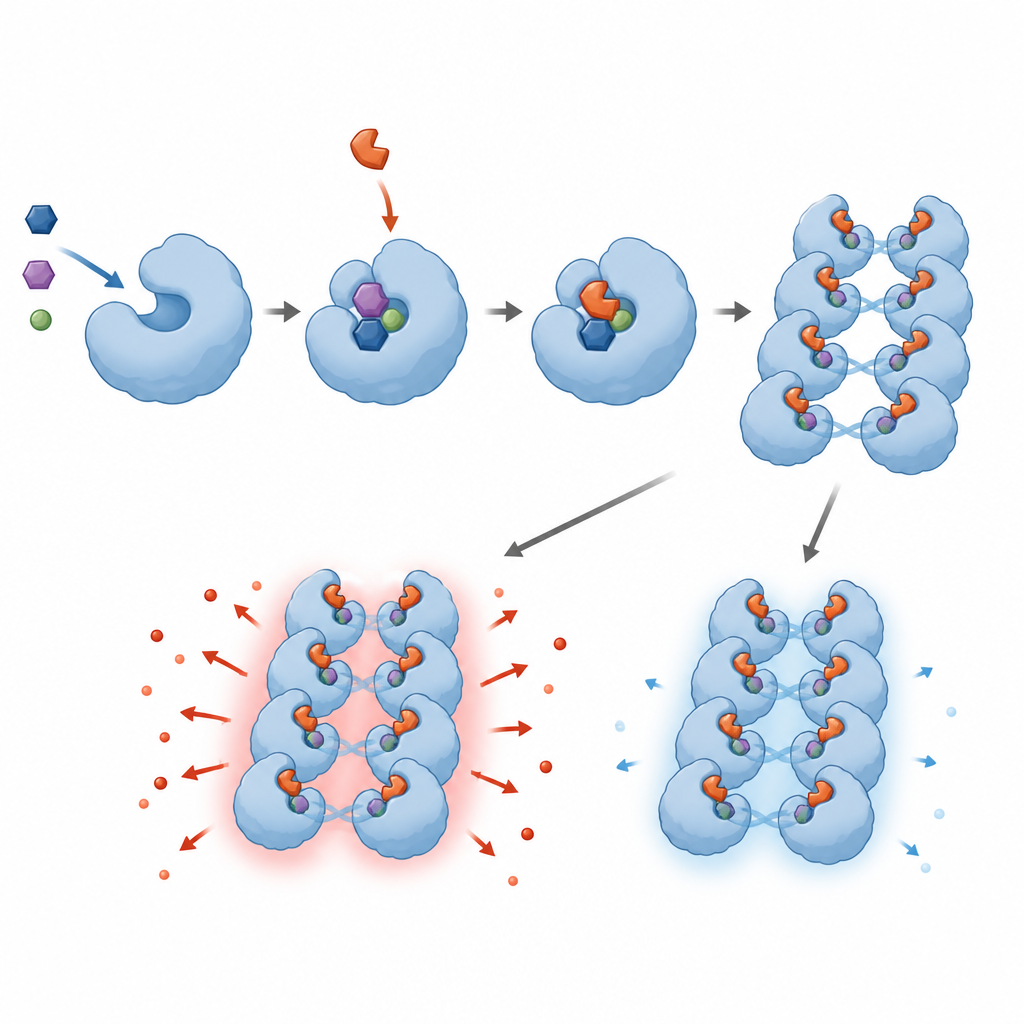
Die Forschenden untersuchten 1,8 Millionen kleine Moleküle, um solche zu finden, die humanes SARM1 im Reagenzglas blockieren. Sie entdeckten eine Familie von Verbindungen, sogenannte Base‑Exchange‑Inhibitoren, die direkt mit dem aktiven Zentrum von SARM1 interagieren und vom Enzym in NAD‑ähnliche Addukte umgewandelt werden, die dort steckenbleiben. Diese Addukte schalten SARM1s Fähigkeit, NAD+ abzubauen, in biochemischen Tests aus und schützten nervenähnliche Zellen sowie sensorische Neuronen von Nagetieren vor Schäden durch Chemotherapeutika oder ein Neurotoxin. Das Team verfeinerte die chemischen Strukturen, um Wirksamkeit, Löslichkeit und Sicherheit zu verbessern, und bestätigte die Bindungsweise der Verbindungen an SARM1 mit fortgeschrittenen Methoden wie Kryo‑Elektronenmikroskopie und Wasserstoff–Deuterium‑Austausch.
Versteckte Wendung bei niedrigen Wirkstoffspiegeln
Als das Team über die Standard‑Screening‑Bedingungen hinausging, entdeckte es ein unerwartetes und beunruhigendes Verhalten. Werden Base‑Exchange‑Inhibitoren in Dosen eingesetzt, die zu niedrig sind, um SARM1 vollständig zu blockieren, werden sie nicht einfach unwirksam; sie machen SARM1 tatsächlich aktiver. In Kulturversuchen mit Nagetier‑ und Humanneuronen führten diese subinhibitorischen Konzentrationen zu schnellerer Axondegeneration, begleitet von erhöhten Pegeln eines SARM1‑abhängig erzeugten Signalstoffs. In gentechnisch veränderten Zelllinien beschleunigten niedrige Dosen derselben Wirkstoffe den NAD+-Verlust statt ihn zu verhindern. Detaillierte strukturelle und biophysikalische Analysen deuten darauf hin, dass die inhibitor‑abgeleiteten Addukte SARM1‑Einheiten dazu anregen, zu längeren Assemblierungen zu verklumpen, wodurch die Gesamtaktivität des Enzyms steigt, sofern nicht nahezu jede Bindungsstelle besetzt ist.
Was im lebenden Tier passiert
Um zu prüfen, ob dieses Paradoxon im Organismus auftritt, testeten die Forschenden einen fortgeschrittenen Base‑Exchange‑Inhibitor, der bereits von anderen Gruppen verwendet wurde, in einem Mausmodell für Ischiasnervverletzung. Anfangs zeigten die behandelten Mäuse niedrigere Werte eines Blutmarkers für Axonschäden, was auf einen vorübergehenden Schutz hindeutete, solange die Wirkstoffspiegel hoch waren. Als die Verbindung jedoch aus dem Körper eliminiert wurde, stiegen die Markerwerte wieder an und übertrafen sogar die der unbehandelten Tiere, was zu einem verzögerten, aber letztlich stärkeren Nervenschaden passt. In Verbindung mit ähnlichen Berichten aus anderen Laboren deuten diese Ergebnisse auf ein reales Risiko hin, dass partielle Dosierung dieser Wirkstoffklasse Nervenschäden verschlimmern könnte statt sie zu lindern.
Was das für künftige nervenschützende Medikamente bedeutet
Die Arbeit zeigt, dass SARM1 weiterhin ein attraktives Ziel zur Verhinderung von Axonverlust bleibt, Base‑Exchange‑Inhibitoren aber wahrscheinlich keine sicheren Medikamente sind. Weil sie vom SARM1 selbst hergestellt und gebunden werden, können sie wie molekulare Klebstoffe wirken, die das Zielprotein zunächst stilllegen und dann überaktivieren — sofern die Wirkstoffspiegel nicht dauerhaft hoch gehalten werden. Die Autoren stoppten daher die Entwicklung dieser Verbindungsfamilie und plädieren dafür, zukünftige Bemühungen auf andere Wege der SARM1‑Modulation zu konzentrieren, etwa durch Zielsetzung seiner regulatorischen Domänen oder durch genetische Ansätze. Ihre Erfahrungen unterstreichen, wie wichtig es ist, leistungsfähige Screening‑Werkzeuge mit sorgfältigen mechanistischen Studien zu koppeln, bevor nervenschützende Medikamente in die Klinik gelangen.
Zitation: Lundbäck, T., Chandrasekar, V., Gu, C. et al. The rise and fall of SARM1 base-exchange inhibitors. Commun Chem 9, 183 (2026). https://doi.org/10.1038/s42004-026-02074-8
Schlüsselwörter: SARM1, Axondegeneration, durch Chemotherapie induzierte Neuropathie, Neuroprotektion, NAD‑Stoffwechsel