Clear Sky Science · it
Mirare l’asse ATP11B‑YAP ripara la funzione mitocondriale e inibisce la ferroptosi neuronale per attenuare il declino cognitivo legato all’età
Perché è importante con l’avanzare dell’età
Con l’aumentare dell’aspettativa di vita, molte persone sono meno preoccupate di aggiungere anni alla vita e più intenzionate a mantenere la lucidità mentale. Questo studio indaga perché il cervello invecchiato diventa vulnerabile alla perdita di memoria e a malattie come l’Alzheimer, e individua un unico “interruttore” molecolare—chiamato ATP11B—that contribuisce a proteggere le cellule cerebrali da una forma lenta di danno guidata dal ferro. Comprendendo come questo interruttore venga compromesso con l’età, i ricercatori delineano una possibile nuova strategia per rallentare o addirittura invertire il declino cognitivo legato all’età.
Barriere che perdono e accumulo di ferro nel cervello invecchiato
Il cervello è normalmente protetto da un sistema filtrante stretto—la barriera sangue‑cervello e strutture correlate—che controllano con precisione ciò che entra dal flusso sanguigno. Nei topi anziani, e nei topi privi geneticamente di ATP11B, questa barriera diventa più permeabile. Il gruppo ha mostrato che questi animali lasciavano passare più piccole molecole nei tessuti cerebrali e i livelli di ferro libero aumentavano bruscamente in regioni chiave, in particolare nell’ippocampo, essenziale per apprendimento e memoria. I test comportamentali hanno rivelato che i topi senza ATP11B invecchiavano più rapidamente, mostravano interazioni sociali peggiori, si muovevano con meno sicurezza e ottenevano risultati inferiori nei labirinti e nei compiti di memoria, imitando i problemi cognitivi legati all’età nell’uomo.
Come le cellule di supporto gestiscono male il ferro
Utilizzando sequenziamento a singola cellula e mappature spaziali, i ricercatori hanno esaminato quali cellule cerebrali fossero maggiormente colpite dalla perdita di ATP11B. Si sono concentrati sulle cellule ependimali, che rivestono gli spazi pieni di liquido del cervello e aiutano a trasferire nutrienti e ioni tra il fluido e il tessuto cerebrale. Nei topi carenti di ATP11B, l’equilibrio tra sottotipi di cellule ependimali si è spostato verso quelle che favoriscono il trasporto di ioni metallici, e queste cellule sembravano migrare più vicino all’ippocampo. Le analisi geniche suggerivano che questo cambiamento promuoveva una consegna anomala di ferro ai neuroni vicini. I marcatori del metabolismo del ferro e della regolazione della memoria in queste regioni risultavano alterati, e i primi segni di invecchiamento cellulare comparivano inizialmente nelle cellule ependimali e poi nei neuroni, suggerendo che il cattivo controllo del ferro da parte delle cellule di supporto può danneggiare i neuroni a distanza.
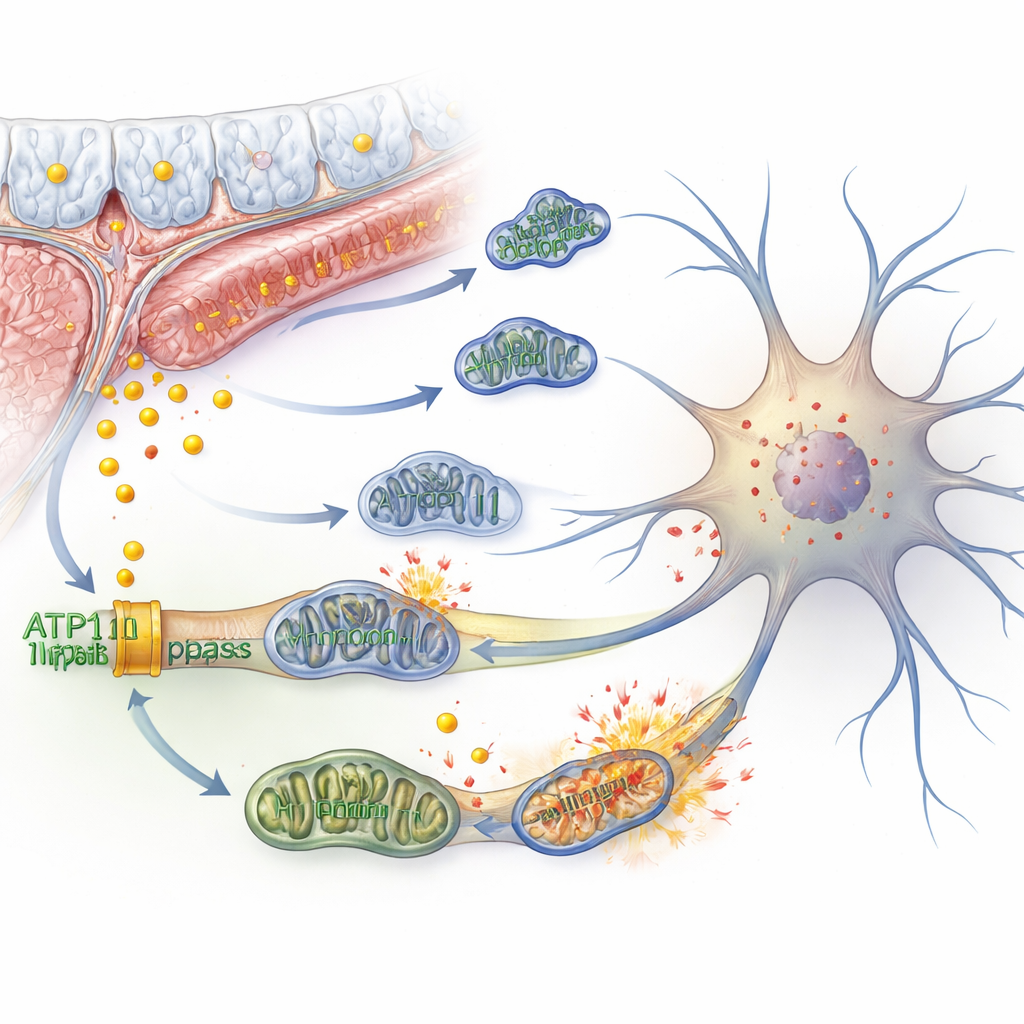
Ferro, centrali elettriche danneggiate e una morte cellulare infuocata
All’interno dei neuroni, il ferro in eccesso può innescare una reazione a catena chiamata ferroptosi—un tipo di morte cellulare promossa dal danno ossidativo ai componenti lipidici della membrana. Gli autori hanno mostrato che la riduzione di ATP11B in cellule nervose di origine umana aumentava il ferro libero, diminuiva la capacità di immagazzinamento del ferro e aumentava i marcatori di ferroptosi. Allo stesso tempo, i mitocondri delle cellule—le piccole centrali che generano energia—diventavano strutturalmente alterati, perdevano il potenziale di membrana e consumavano meno ossigeno. Le cellule cercavano di compensare incrementando la glicolisi e producendo più lattato, ma questo comportava una maggiore presenza di molecole reattive che danneggiavano ulteriormente le membrane. I mitocondri si frammentavano invece di fondersi, i processi di pulizia cellulare erano iperattivati e comparivano segnali di invecchiamento cellulare e aumento di specie reattive dell’ossigeno, tutti fattori che contribuivano a indebolire la trasmissione elettrica tra i neuroni.
Un asse di controllo molecolare nel nucleo
Lo studio è andato oltre per chiedersi come ATP11B regoli così tanti eventi a valle. Esaminando come il DNA è impacchettato e letto nei neuroni, il gruppo ha scoperto che la perdita di ATP11B rende la cromatina—il materiale che avvolge il DNA—meno accessibile in siti specifici. Questo ha colpito in particolare le regioni di legame per un fattore di trascrizione chiamato KLF4, che normalmente contribuisce a mantenere l’espressione di geni necessari per una funzione mitocondriale sana. Allo stesso tempo, una via di controllo della crescita nota come Hippo divenne meno attiva, permettendo a una proteina chiamata YAP di entrare nel nucleo e formare un complesso che attiva geni collegati alla ferroptosi e all’invecchiamento. Il maggiore lattato prodotto da mitocondri sotto stress influiva anche sui segni chimici degli istoni (istone lattilazione), aumentando ulteriormente l’espressione di geni chiave pro‑invecchiamento e pro‑ferroptosi. Insieme, questi cambiamenti nucleari intrappolarono i neuroni in un ciclo auto‑rinforzante di stress da ferro e fallimento mitocondriale.
Riaccendere l’interruttore
In modo incoraggiante, il danno non era del tutto irreversibile. Quando i ricercatori hanno somministrato ATP11B in eccesso a topi anziani usando un vettore virale, molti problemi tipici dell’età sono migliorati. I topi trattati camminavano con passi più lunghi e stabili, mantenevano meglio l’equilibrio, esploravano più volentieri ambienti nuovi e ottenevano risultati significativamente migliori nei test di memoria. Al microscopio, i loro neuroni mostravano rami più lunghi e spine più complesse, caratteristiche strutturali associate a sinapsi più forti. I marcatori molecolari indicavano che il danno lipidico indotto dal ferro e lo stress mitocondriale erano ridotti. In termini semplici, ripristinare ATP11B ha aiutato a riequilibrare la gestione del ferro e la salute mitocondriale, attenuando la pressione che spinge i neuroni invecchiati verso la morte e il declino cognitivo.
Cosa significa per la salute del cervello
Per un lettore non specialista, questo lavoro suggerisce che parte dell’invecchiamento cerebrale potrebbe ridursi a un guasto specifico nel modo in cui le cellule gestiscono il ferro, la produzione di energia e il controllo genico. ATP11B agisce come un coordinatore centrale: quando viene perso, il ferro si accumula silenziosamente, le centrali cellulari vacillano e un processo distruttivo, simile alla ruggine, erode lentamente i circuiti della memoria. Quando ATP11B viene ripristinato, almeno nei topi, molte di queste tendenze possono essere invertite. Trasformare questa intuizione in terapie sicure per l’uomo richiederà però grandi progressi nelle tecnologie di somministrazione genica e test a lungo termine; nondimeno, lo studio evidenzia un nuovo bersaglio promettente per preservare capacità cognitive, memoria e autonomia in una popolazione che invecchia.
Citazione: Qi, W., Liu, Q., Dong, N. et al. Targeting ATP11B-YAP axis repairs mitochondrial function and inhibits neuronal ferroptosis to attenuate age-related cognitive decline. Sig Transduct Target Ther 11, 141 (2026). https://doi.org/10.1038/s41392-026-02652-1
Parole chiave: invecchiamento cerebrale, eccesso di ferro, disfunzione mitocondriale, ferroptosi neuronale, declino cognitivo