Clear Sky Science · en
Targeting ATP11B-YAP axis repairs mitochondrial function and inhibits neuronal ferroptosis to attenuate age-related cognitive decline
Why This Matters for Growing Older
As people live longer, many worry less about adding years to life and more about keeping those years mentally sharp. This study explores why the aging brain becomes vulnerable to memory loss and diseases like Alzheimer’s, and pinpoints a single molecular "switch"—called ATP11B—that helps protect brain cells from a slow, iron‑driven form of damage. By understanding how this switch fails with age, the researchers outline a possible new way to slow or even reverse age‑related cognitive decline.
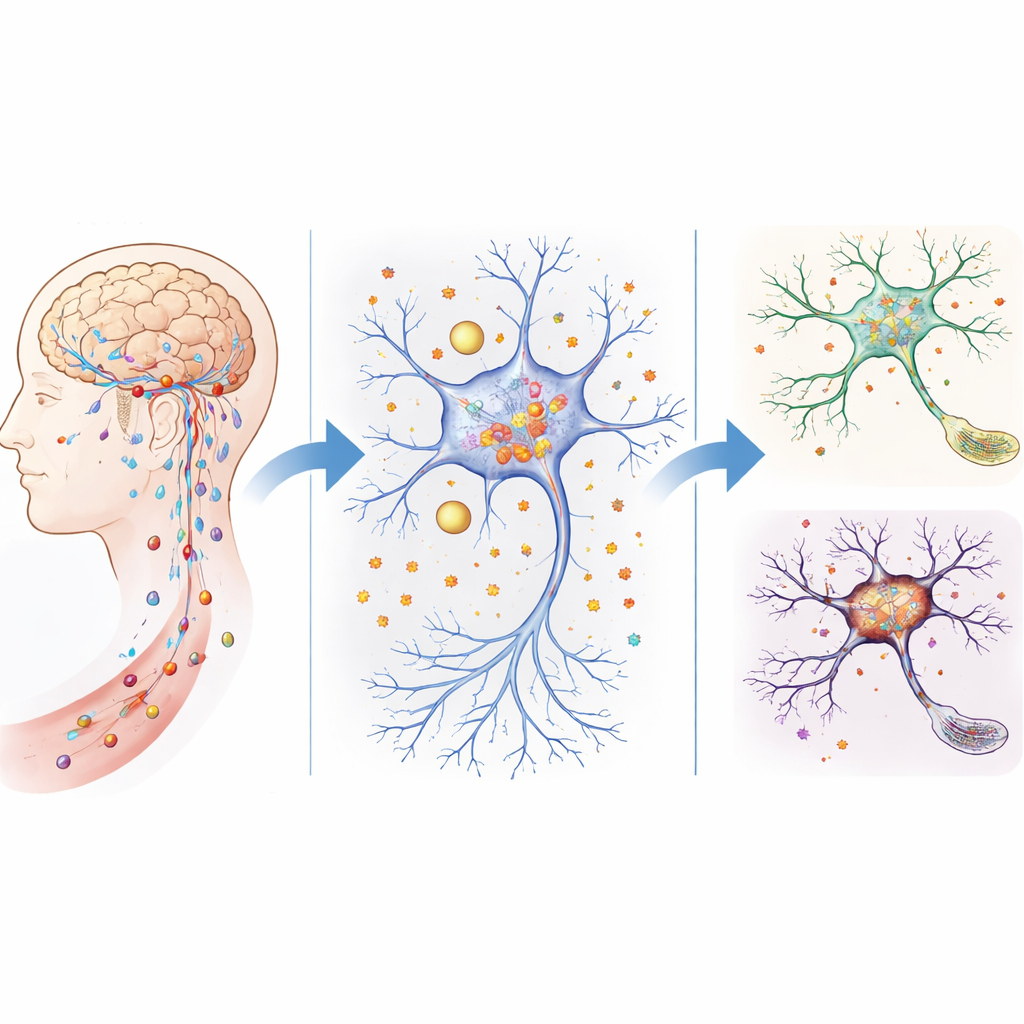
Leaky Defenses and Iron Buildup in the Aging Brain
The brain is normally guarded by a tight filter system—the blood–brain barrier and related structures—that carefully control what enters from the bloodstream. In older mice, and in mice genetically lacking ATP11B, this barrier becomes leakier. The team showed that these mice allowed more small molecules to seep into brain tissue, and the levels of free iron rose sharply in key regions, especially the hippocampus, which is crucial for learning and memory. Behavior tests revealed that mice without ATP11B aged faster, showed poorer social interaction, moved less confidently, and performed worse in maze and memory tasks, mimicking human age‑related cognitive problems.
How Support Cells Mismanage Iron
Using single‑cell sequencing and spatial mapping, the researchers examined which brain cells were most affected by the loss of ATP11B. They focused on ependymal cells, which line the brain’s fluid‑filled spaces and help shuttle nutrients and ions between the fluid and brain tissue. In ATP11B‑deficient mice, the balance of ependymal cell subtypes shifted toward those that favor metal ion transport, and these cells appeared to migrate closer to the hippocampus. Gene analyses suggested that this change promoted abnormal iron delivery to nearby neurons. Markers of iron handling and memory regulation in these regions were altered, and early signs of cellular aging appeared first in ependymal cells and then in neurons, implying that mismanaged iron from support cells may drive neuron damage from a distance.
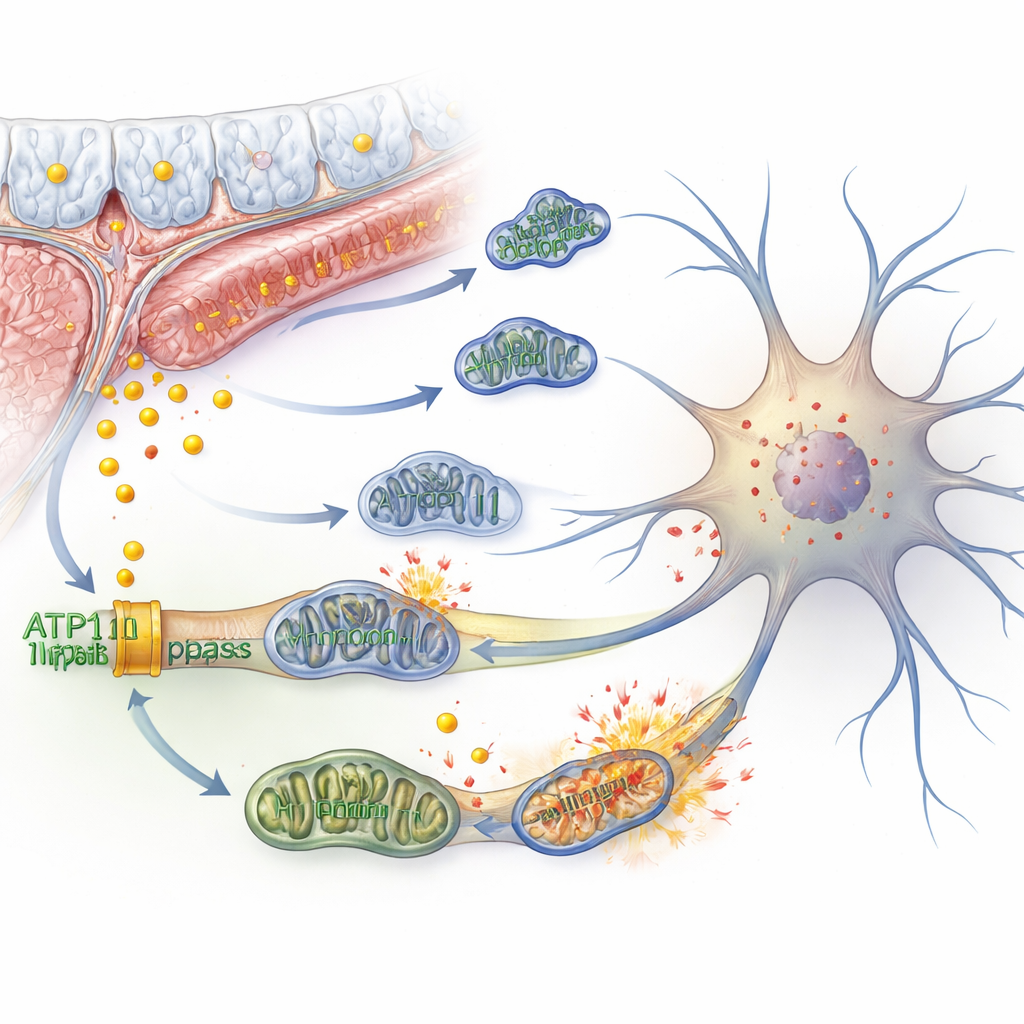
Iron, Broken Powerhouses, and a Fiery Cell Death
Inside neurons, excess iron can spark a chain reaction called ferroptosis—a type of cell death driven by oxidative damage to fatty components of the cell membrane. The authors showed that knocking down ATP11B in human‑derived nerve cells increased free iron, reduced iron storage capacity, and boosted markers of ferroptosis. At the same time, the cells’ mitochondria—the tiny power plants that generate energy—became structurally distorted, lost their membrane potential, and consumed less oxygen. The cells tried to compensate by ramping up sugar‑burning and producing more lactate, but this came with more reactive molecules that further damaged membranes. Mitochondria fragmented instead of fusing, cleanup processes were over‑activated, and signs of cellular aging and rising reactive oxygen levels appeared, all contributing to weaker electrical signaling between neurons.
A Molecular Control Axis in the Nucleus
The study went deeper to ask how ATP11B controls so many downstream events. By examining how DNA is packaged and read in neurons, the team found that ATP11B loss makes chromatin—the material that wraps DNA—less accessible at specific sites. This especially affected binding regions for a transcription factor called KLF4, which normally helps maintain genes for healthy mitochondrial function. At the same time, a growth‑control pathway known as Hippo became less active, allowing a protein called YAP to move into the nucleus and form a complex that switches on genes linked to ferroptosis and aging. Higher lactate from stressed mitochondria also fed into chemical tags on histones (histone lactylation), further boosting expression of key pro‑aging and pro‑ferroptosis genes. Together, these nuclear changes locked neurons into a self‑reinforcing cycle of iron stress and mitochondrial failure.
Turning the Switch Back On
Encouragingly, the damage was not entirely irreversible. When the researchers delivered extra ATP11B to aged mice using a viral vector, many age‑like problems improved. Treated mice walked with longer, more stable strides, maintained balance better, explored new environments more readily, and performed significantly better in memory tests. Under the microscope, their neurons showed longer branches and more complex spines, structural features linked to stronger synaptic connections. Molecular markers indicated that iron‑driven lipid damage and mitochondrial stress were reduced. In simple terms, restoring ATP11B helped reset iron handling and mitochondrial health, easing the pressure that pushes aging neurons toward death and cognitive decline.
What This Means for Brain Health
To a layperson, this work suggests that part of brain aging may boil down to a specific failure in how cells juggle iron, energy production, and gene control. ATP11B acts as a central coordinator: when it is lost, iron quietly accumulates, brain cell powerhouses falter, and a destructive, rust‑like process slowly erodes memory circuits. When ATP11B is restored, at least in mice, many of these trends can be reversed. While turning this insight into safe therapies for people will require major advances in gene delivery and long‑term testing, the study highlights a promising new target for preserving thinking, memory, and independence in an aging population.
Citation: Qi, W., Liu, Q., Dong, N. et al. Targeting ATP11B-YAP axis repairs mitochondrial function and inhibits neuronal ferroptosis to attenuate age-related cognitive decline. Sig Transduct Target Ther 11, 141 (2026). https://doi.org/10.1038/s41392-026-02652-1
Keywords: brain aging, iron overload, mitochondrial dysfunction, neuronal ferroptosis, cognitive decline