Clear Sky Science · fr
Les défauts neurodéveloppementaux dépendant du dosage du gène MeCP2 apparaissent par activation aberrante de gènes bivalents déterminant le destin cellulaire
Pourquoi ce gène est important pour la santé cérébrale
Le gène MECP2 est célèbre car un déficit en sa protéine provoque le syndrome de Rett, tandis qu’un excès conduit au syndrome de duplication de MECP2. Ces deux affections entraînent de graves problèmes comme une déficience intellectuelle, des crises d’épilepsie et des troubles moteurs. Cette étude pose une question apparemment simple mais aux grandes implications pour les thérapies géniques à venir : un excès de MeCP2 est-il toujours dangereux, ou le moment et le type cellulaire où il survient déterminent-ils si le cerveau est lésé ou épargné ?
Quand un surplus de MeCP2 apparaît trop tôt
Les chercheurs ont comparé ce qui se passe lorsque MeCP2 est surproduit dans les « progéniteurs neuraux » immatures par rapport à des neurones pleinement formés, en utilisant des cellules de souris et humaines. Les progéniteurs neuraux sont les cellules en division du cerveau en développement qui donneront ensuite naissance aux neurones. Lorsque l’équipe a augmenté MeCP2 dans ces progéniteurs, l’activité génétique des cellules a changé de façon spectaculaire : des milliers de gènes sont devenus plus ou moins actifs, avec un fort biais vers l’activation de gènes qui poussent les cellules à devenir des neurones. En culture et dans des cerveaux de souris en développement, les progéniteurs avec un excès de MeCP2 se sont divisés moins et se sont convertis en neurones plus tôt et plus rapidement que la normale, modifiant le rythme du développement cérébral.
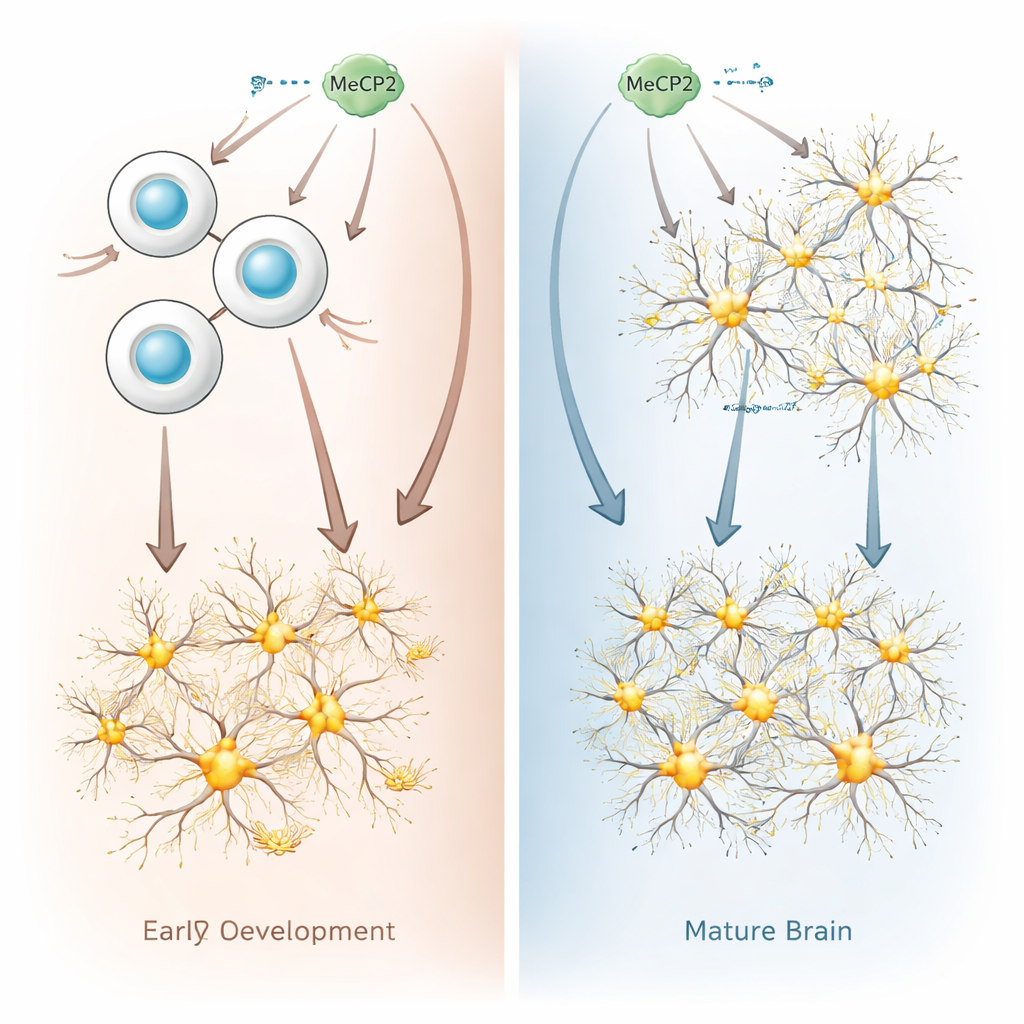
Pourquoi les neurones matures tolèrent mieux l’excès de MeCP2
En net contraste, lorsque la même quantité de MeCP2 a été ajoutée à des neurones matures, les effets furent étonnamment faibles. Seules quelques centaines de gènes ont modifié leur activité, et la plupart de ces changements étaient modestes. Les chercheurs n’ont également trouvé que peu de preuves d’une altération globale du empaquetage de l’ADN dans ces neurones. Chez des souris vivantes, augmenter MeCP2 dans les progéniteurs embryonnaires a produit des neurones adultes aux signaux électriques excitateurs renforcés, en écho aux observations dans les modèles du syndrome de duplication. Mais augmenter MeCP2 directement dans le cerveau adulte n’a pas modifié le comportement électrique des neurones. Ensemble, ces résultats montrent que les neurones matures tolèrent beaucoup mieux une augmentation de MeCP2 que les progéniteurs en développement.
Comment MeCP2 choisit ses sites sur l’ADN
Pour comprendre pourquoi le type cellulaire compte autant, l’équipe a cartographié précisément où se fixent à la fois MeCP2 normal et excédentaire sur l’ADN. Dans les progéniteurs comme dans les neurones, MeCP2 s’est focalisé sur des tronçons d’ADN riches en « îlots CpG » près des sites de démarrage des gènes — des régions qui aident à contrôler si les gènes sont activés ou non. La protéine normale et la protéine en excès ont choisi essentiellement le même ensemble de cibles, en particulier des gènes impliqués dans la construction et le raffinage des circuits neuronaux. La différence clé résidait dans l’occupation de ces sites. Dans les neurones, où MeCP2 est déjà naturellement abondant, ces sites étaient presque saturés, laissant peu de place pour la protéine supplémentaire, qui se liait faiblement et était dégradée plus rapidement. Dans les progéniteurs, où les niveaux de MeCP2 sont normalement bas, la protéine ajoutée pouvait se lier beaucoup plus fortement et largement à ces régions régulatrices.
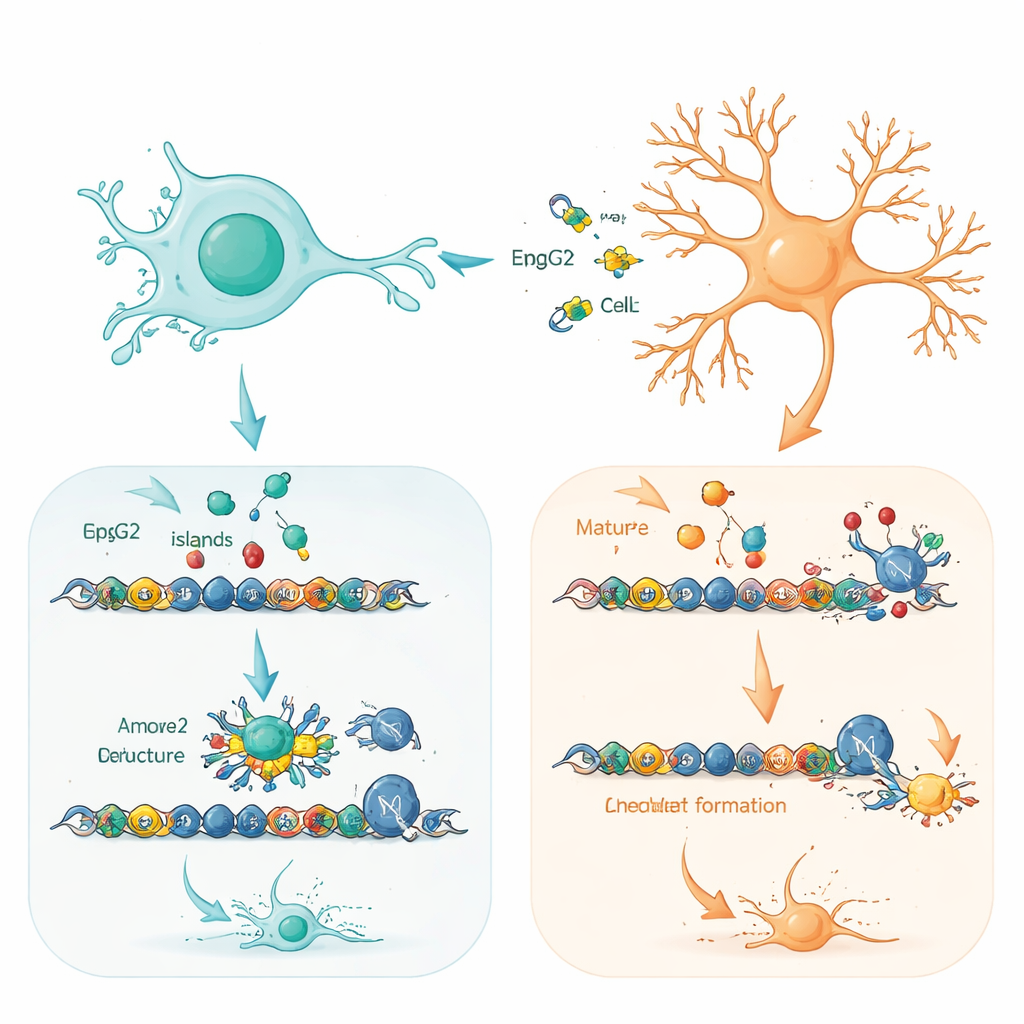
Préparer les gènes déterminant le destin dans les cellules cérébrales jeunes
Une découverte particulièrement marquante est que beaucoup des gènes les plus affectés chez les progéniteurs se trouvent dans un état « prêt » : ils portent à la fois des marques chimiques activatrices et répressives sur leurs régions régulatrices et sont prêts à s’activer au cours du développement. Ces gènes bivalents contrôlent souvent des décisions clés sur les types de neurones produits et leur calendrier. Les auteurs montrent qu’un excès de MeCP2 aide à recruter une machine puissante de remodelage de la chromatine, le complexe SWI/SNF, à ces sites prêts. Ce partenariat fait pencher la balance vers l’activation, déclenchant l’ensemble des programmes de différenciation neuronale plus tôt qu’ils ne devraient l’être. Des modifications subtiles du paysage plus large de l’emballage de l’ADN confirment ce tableau : des régions liées au contrôle du cycle cellulaire et à la maturation neuronale sont devenues légèrement plus ouvertes dans les progéniteurs avec un excès de MeCP2.
Ce que cela implique pour la thérapie génique et les maladies cérébrales
Pour les familles et les cliniciens inquiets que les thérapies géniques ciblant MeCP2 puissent dépasser la cible et nuire au cerveau, ce travail offre une prudente consolation. L’étude suggère que des augmentations modérées de MeCP2 dans les neurones matures — même de trois à quatre fois — sont étonnamment bien tolérées, parce que les sites de liaison sont déjà occupés et que la protéine excédentaire est rapidement éliminée. Le véritable danger apparaît lorsque MeCP2 est élevé tôt dans le développement, dans des cellules progénitrices dont les gènes déterminant le destin sont encore prêts et très sensibles. Dans ce contexte, un surplus de MeCP2 peut activer prématurément des programmes de développement, modifier la manière et le moment de production des neurones, et au final remodeler les connexions cérébrales d’une façon qui peut contribuer à l’épilepsie et à d’autres symptômes observés dans le syndrome de duplication de MECP2. Plus largement, ces résultats soulignent un principe probablement partagé par de nombreux régulateurs de la chromatine : le dosage génique n’est pas intrinsèquement toxique, mais son impact dépend crucialement du moment du développement et du type cellulaire où survient le déséquilibre.
Citation: Luoni, M., Kubacki, M., Giannelli, S.G. et al. MeCP2 gene dosage-dependent neurodevelopmentally restricted defects arise by aberrant activation of cell fate-determining bivalent genes. Nat Commun 17, 3225 (2026). https://doi.org/10.1038/s41467-026-71432-w
Mots-clés: MeCP2, neurodéveloppement, dosage génique, épigénétique, thérapie génique