Clear Sky Science · de
Dosierungsabhängige, neuroentwicklungsbedingt begrenzte Defekte des MeCP2-Gens entstehen durch fehlgeleitete Aktivierung von schicksalsbestimmenden bivalenten Genen
Warum dieses Gen für die Gesundheit des Gehirns wichtig ist
Das MECP2-Gen ist bekannt, weil ein Mangel seines Proteins das Rett-Syndrom verursacht, während ein Zuviel zur MeCP2-Duplikationssyndrom führt. Beide Zustände bringen schwere Probleme wie geistige Behinderung, Anfälle und Bewegungsstörungen mit sich. Diese Studie stellt eine auf den ersten Blick einfache, aber weitreichende Frage für zukünftige Gentherapien: Ist zusätzliches MeCP2 immer schädlich, oder bestimmen Zeitpunkt und Zelltyp, in denen es auftritt, ob das Gehirn geschädigt wird oder verschont bleibt?
Wenn zusätzliches MeCP2 zu früh auftaucht
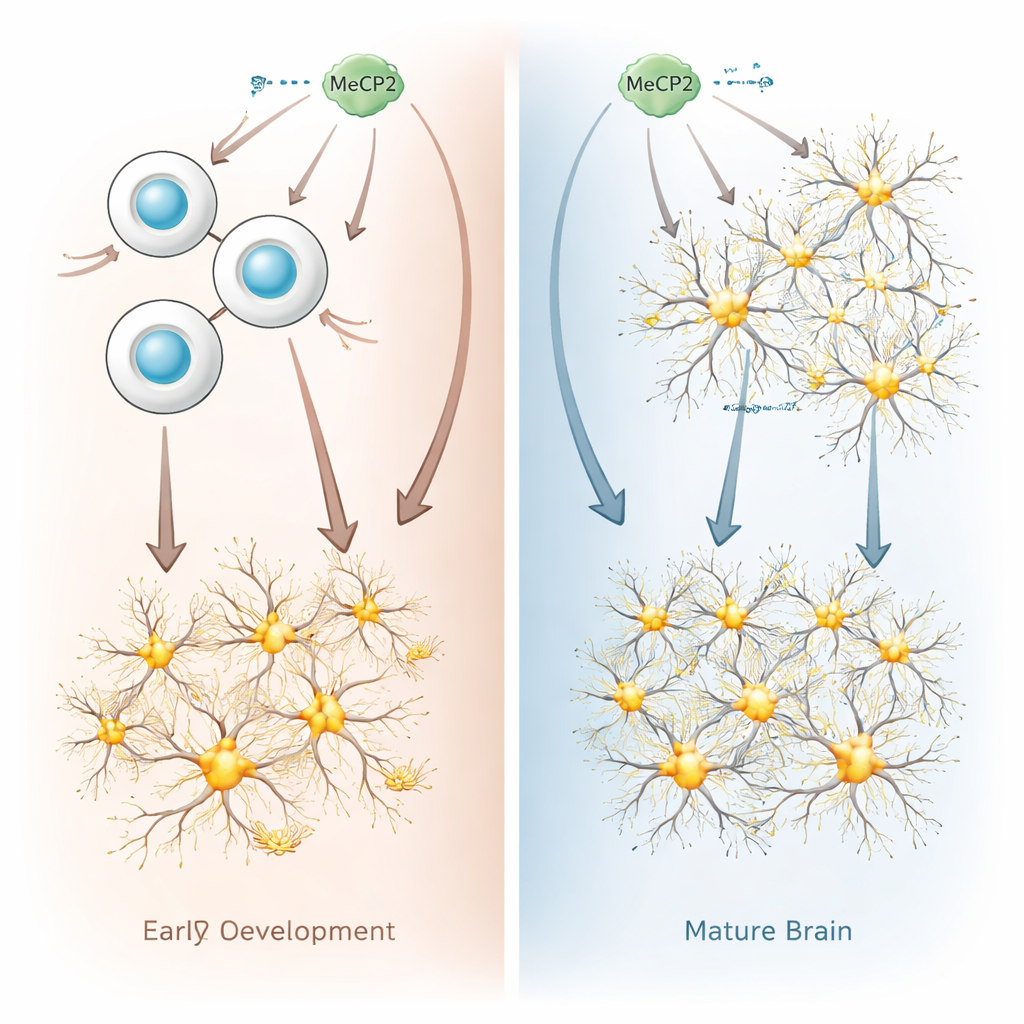
Die Forscher verglichen, was passiert, wenn MeCP2 in unreifen „neuralen Vorläufer“-Zellen im Vergleich zu vollständig ausgebildeten Neuronen überproduziert wird, und zwar sowohl in Maus- als auch in menschlichen Zellen. Neurale Vorläufer sind die teilenden Zellen im sich entwickelnden Gehirn, die später Neuronen hervorbringen. Als das Team MeCP2 in diesen Vorläufern erhöhte, veränderte sich die Genaktivität dramatisch: Tausende Gene wurden stärker oder schwächer aktiv, mit einer deutlichen Tendenz zur Aktivierung von Genen, die Zellen in Richtung Neuronalschicksal treiben. In Zellkulturen und in sich entwickelnden Mausgehirnen teilten sich Vorläufer mit zu viel MeCP2 weniger und differenzierten schneller und früher zu Neuronen als normal, wodurch das Tempo der Gehirnentwicklung verschoben wurde.
Warum reife Neuronen zusätzliches MeCP2 gelassener wegstecken
Im scharfen Kontrast dazu wirkten sich dieselben MeCP2-Mengen in reifen Neuronen überraschend schwach aus. Nur einige hundert Gene änderten ihre Aktivität, und die meisten dieser Veränderungen waren klein. Die Forscher fanden auch kaum Hinweise darauf, dass die allgemeine DNA-Verpackung in diesen Neuronen verändert wurde. In lebenden Mäusen führte eine Erhöhung von MeCP2 in embryonalen Vorläufern zu adulten Neuronen mit stärkeren exzitatorischen elektrischen Signalen, was dem entspricht, was in Modellen des Duplikationssyndroms beobachtet wird. Eine Erhöhung von MeCP2 direkt im erwachsenen Gehirn veränderte jedoch nicht das elektrische Verhalten der Neuronen. Insgesamt zeigen diese Ergebnisse, dass reife Neuronen gegenüber erhöhtem MeCP2 deutlich toleranter sind als sich entwickelnde Vorläufer.
Wie MeCP2 seine Stellen auf der DNA auswählt
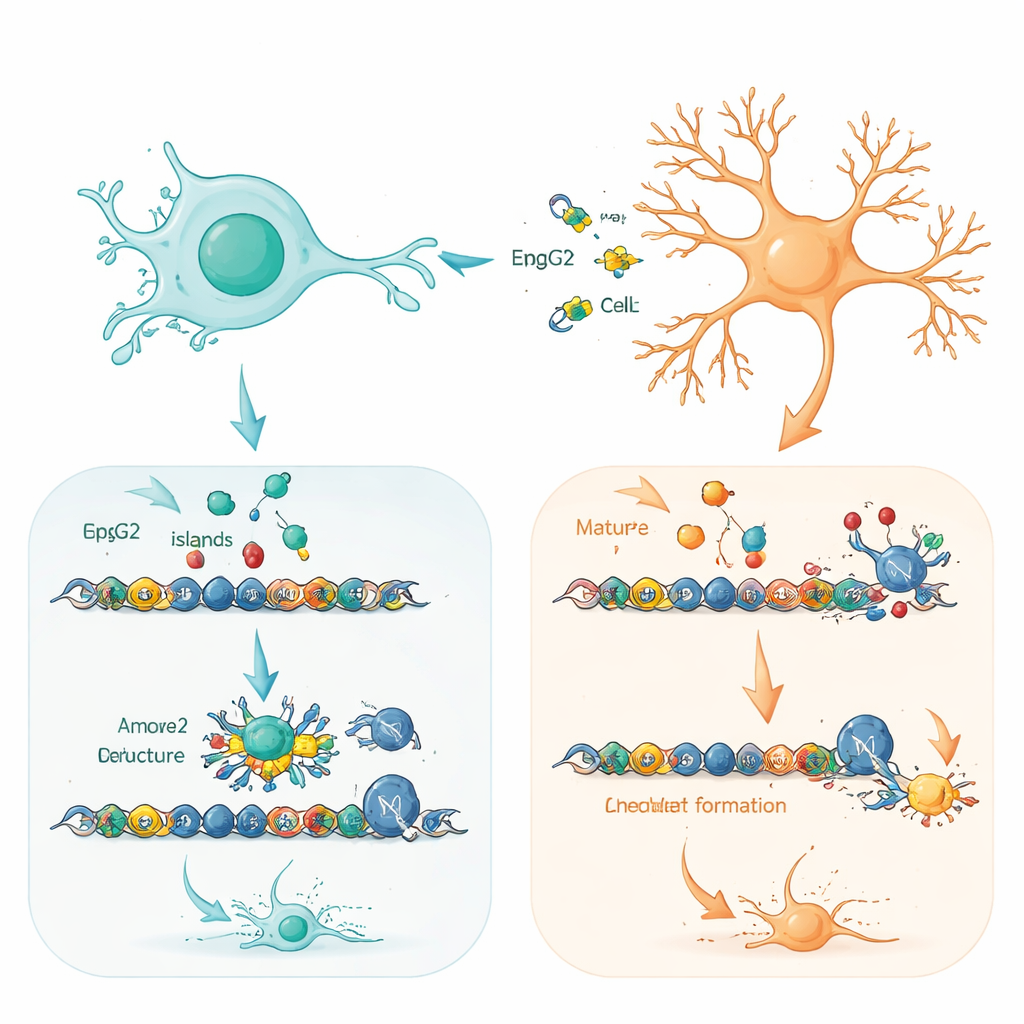
Um zu verstehen, warum der Zelltyp so entscheidend ist, kartierte das Team genau, wo sich sowohl normales als auch zusätzliches MeCP2 auf der DNA befindet. In Vorläufern und Neuronen konzentrierte sich MeCP2 auf DNA-Abschnitte, die reich an „CpG-Inseln“ in der Nähe von Genstartpunkten sind — Regionen, die steuern, ob Gene an- oder ausgeschaltet sind. Das normale und das zusätzliche Protein wählten im Wesentlichen dasselbe Set an Zielen, insbesondere Gene, die am Aufbau und der Feinabstimmung neuronaler Schaltkreise beteiligt sind. Der entscheidende Unterschied lag in der Besetzungsdichte dieser Stellen. In Neuronen, in denen MeCP2 natürlicherweise bereits reichlich vorhanden ist, waren diese Stellen nahezu gesättigt, so dass für zusätzliches Protein kaum Raum blieb; dieses band schwächer und wurde schneller abgebaut. In Vorläufern, in denen MeCP2 normalerweise gering ist, konnte das hinzugefügte Protein deutlich stärker und weiterreichender an diesen regulatorischen Regionen binden.
Voraussetzungsschaffung für schicksalsentscheidende Gene in jungen Gehirnzellen
Eine besonders bemerkenswerte Beobachtung war, dass viele der am stärksten betroffenen Gene in Vorläufern in einem „bereiten“ Zustand vorliegen: Sie tragen an ihren regulatorischen Regionen sowohl aktivierende als auch repressive chemische Markierungen und sind bereit, während der Entwicklung angeschaltet zu werden. Diese bivalenten Gene steuern häufig wichtige Entscheidungen darüber, welche Neuronentypen wann entstehen. Die Autoren zeigen, dass überschüssiges MeCP2 dabei hilft, einen mächtigen DNA-Verpackungsapparat, den SWI/SNF-Komplex, an diese bereiten Stellen zu rekrutieren. Diese Partnerschaft verlagert das Gleichgewicht in Richtung Aktivierung und schaltet ganze Programme der neuronalen Differenzierung früher an als vorgesehen. Dezente Verschiebungen in der breiteren DNA-Packungslandschaft stützen dieses Bild: Regionen, die mit Zellzykluskontrolle und neuronaler Reifung verknüpft sind, wurden in Vorläufern mit überschüssigem MeCP2 leicht zugänglicher.
Was das für Gentherapie und Hirnerkrankungen bedeutet
Für Familien und Klinikern, die befürchten, dass MeCP2-basierte Gentherapien über das Ziel hinausschießen und dem Gehirn schaden könnten, bietet diese Arbeit vorsichtige Beruhigung. Die Studie legt nahe, dass moderate MeCP2-Erhöhungen in reifen Neuronen — selbst dreifach bis vierfach — überraschend gut vertragen werden, weil Bindungsstellen bereits besetzt sind und überschüssiges Protein rasch entfernt wird. Die eigentliche Gefahr besteht offenbar, wenn MeCP2 früh in der Entwicklung in Vorläuferzellen erhöht ist, deren schicksalsentscheidende Gene noch bereit und hochsensibel sind. In diesem Kontext kann überschüssiges MeCP2 Entwicklungsprogramme vorzeitig aktivieren, verändern, wie und wann Neuronen entstehen, und letztlich die Verschaltung des Gehirns so beeinflussen, dass dies zu Epilepsie und anderen Symptomen des MECP2-Duplikationssyndroms beitragen kann. Allgemeiner heben die Ergebnisse ein Prinzip hervor, das wahrscheinlich von vielen Chromatin-Regulatoren geteilt wird: Gen-Dosierung ist nicht per se toxisch, aber ihre Auswirkungen hängen entscheidend davon ab, wann in der Entwicklung und in welchem Zelltyp das Ungleichgewicht auftritt.
Zitation: Luoni, M., Kubacki, M., Giannelli, S.G. et al. MeCP2 gene dosage-dependent neurodevelopmentally restricted defects arise by aberrant activation of cell fate-determining bivalent genes. Nat Commun 17, 3225 (2026). https://doi.org/10.1038/s41467-026-71432-w
Schlüsselwörter: MeCP2, Neuroentwicklung, Gen-Dosierung, Epigenetik, Gentherapie