Clear Sky Science · fr
Défluoroallylation radicalaire de composés polyfluorés avec des alcènes via une catalyse synergique photoredox/cobalt
Pourquoi il est important de modifier des molécules riches en fluor
Les composés fortement fluorés sont omniprésents dans la vie moderne, des médicaments et agents de protection des cultures aux revêtements anti‑taches. Leur efficacité provient des liaisons carbone–fluor, parmi les plus fortes en chimie organique, ce qui rend ces composés exceptionnellement stables dans l'organisme et dans l'environnement. Cette même robustesse rend cependant leur modification intentionnelle très difficile. Cette étude présente une voie douce pour rompre une liaison carbone–fluor précise dans de longues chaînes fluorées et la remplacer par une liaison carbone–carbone utile, ce qui peut aider les chimistes à concevoir de meilleurs médicaments et matériaux à partir de ces molécules rétives.
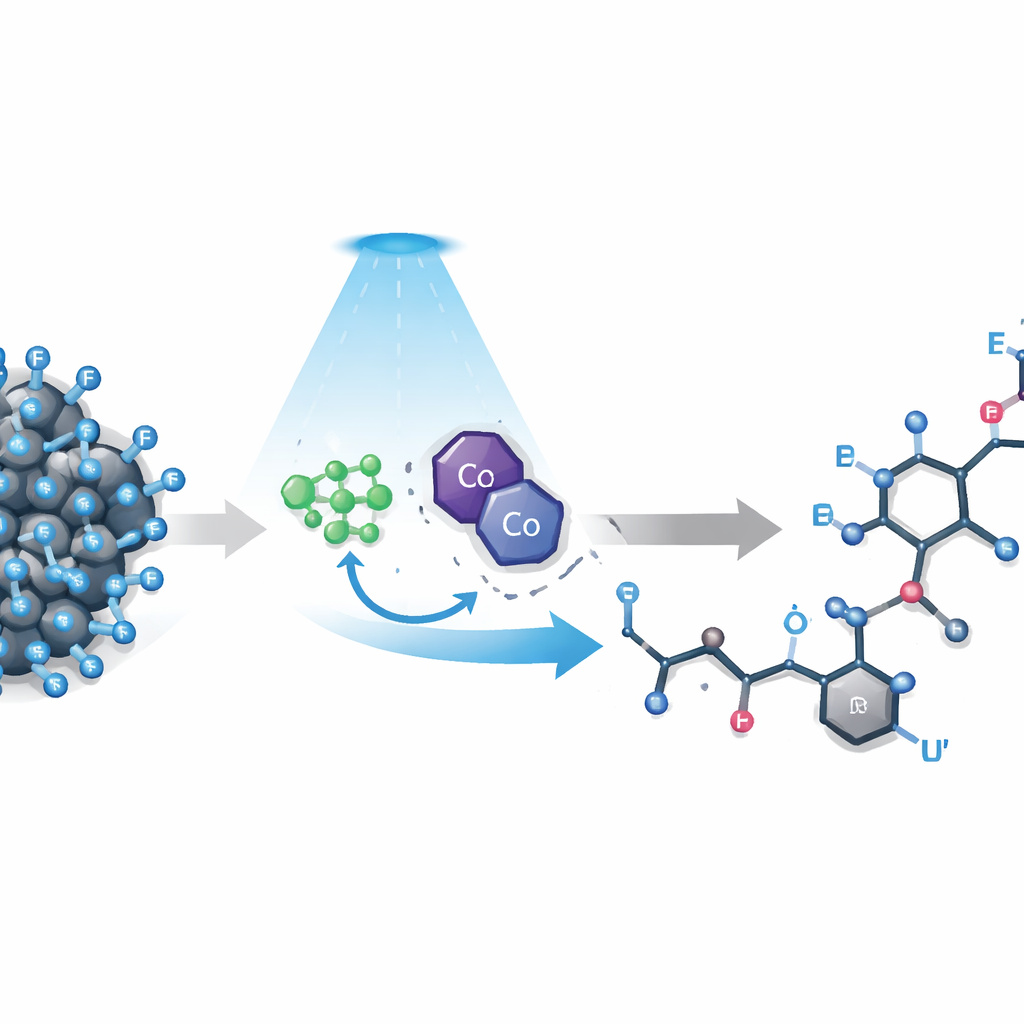
Le défi de couper la liaison la plus forte
Les composés polyfluoroalkyles contiennent des chaînes où de nombreux hydrogènes ont été remplacés par du fluor. Ces chaînes rendent les molécules hydrophobes et chimiquement résistantes, qualités recherchées en technologie et en médecine, mais problématiques lorsque les chimistes veulent ajuster leur comportement. Les méthodes existantes se concentrent souvent sur le groupe fluoré le plus simple, CF3, ou exigent des additifs très réactifs et des partenaires d'accouplement préformés. Les chaînes plus longues, courantes dans les produits réels, sont encore plus difficiles à maîtriser : l'attraction exercée par le fluor déstabilise les intermédiaires réactifs, les atomes de fluor encombrants bloquent l'accès et des flux électroniques inverses peuvent annuler les premières étapes. Par conséquent, les tentatives de rompre une liaison carbone–fluor choisie aboutissent souvent à des mélanges désordonnés ou à un échec total.
Une approche à deux catalyseurs alimentée par la lumière
Les auteurs ont élaboré une stratégie qui utilise deux catalyseurs coopérants et la lumière visible pour dompter ces molécules difficiles. Un complexe d'iridium photosensible cède d'abord un électron unique à un composé polyfluoroalkyl, affaiblissant suffisamment une liaison carbone–fluor pour qu'elle se rompe et libère un fragment fluoré de courte durée de vie connu comme un radical. Parallèlement, un catalyseur au cobalt inspiré de la vitamine B12 oriente l'endroit où un atome d'hydrogène sera finalement ajouté. Un simple réactif à base de bore joue le rôle d'éponge pour le fluorure, piégeant le fluor expulsé et empêchant sa recombinaison ou le transfert électronique inverse. Sous une illumination douce par LED bleue, ce réseau d'assistants conduit la réaction selon une voie productive et hautement sélective.
Construire de nouvelles liaisons avec des alcènes simples
Une fois formés, les radicaux fluorés s'additionnent à des alcènes courants — petites molécules portant une double liaison carbone–carbone — créant de nouvelles liaisons carbone–carbone en une seule étape. Le catalyseur au cobalt transfert ensuite un atome d'hydrogène à une position voisine spécifique, verrouillant un fragment « allyl » tout en préservant la double liaison utile. Sur des dizaines d'essais, la méthode a fonctionné pour une large gamme d'alcènes, y compris des terpènes naturels complexes, et pour de nombreux matériaux de départ riches en fluor présentant des chaînes de trois à dix atomes de carbone ou des groupes amides fluorés. Fait remarquable, même lorsque les alcènes comportaient plusieurs sites allyliques C–H presque identiques, la réaction a choisi une position avec une sélectivité supérieure à 20 pour 1, évitant l'enchevêtrement d'isomères qui complique généralement ce type de chimie.
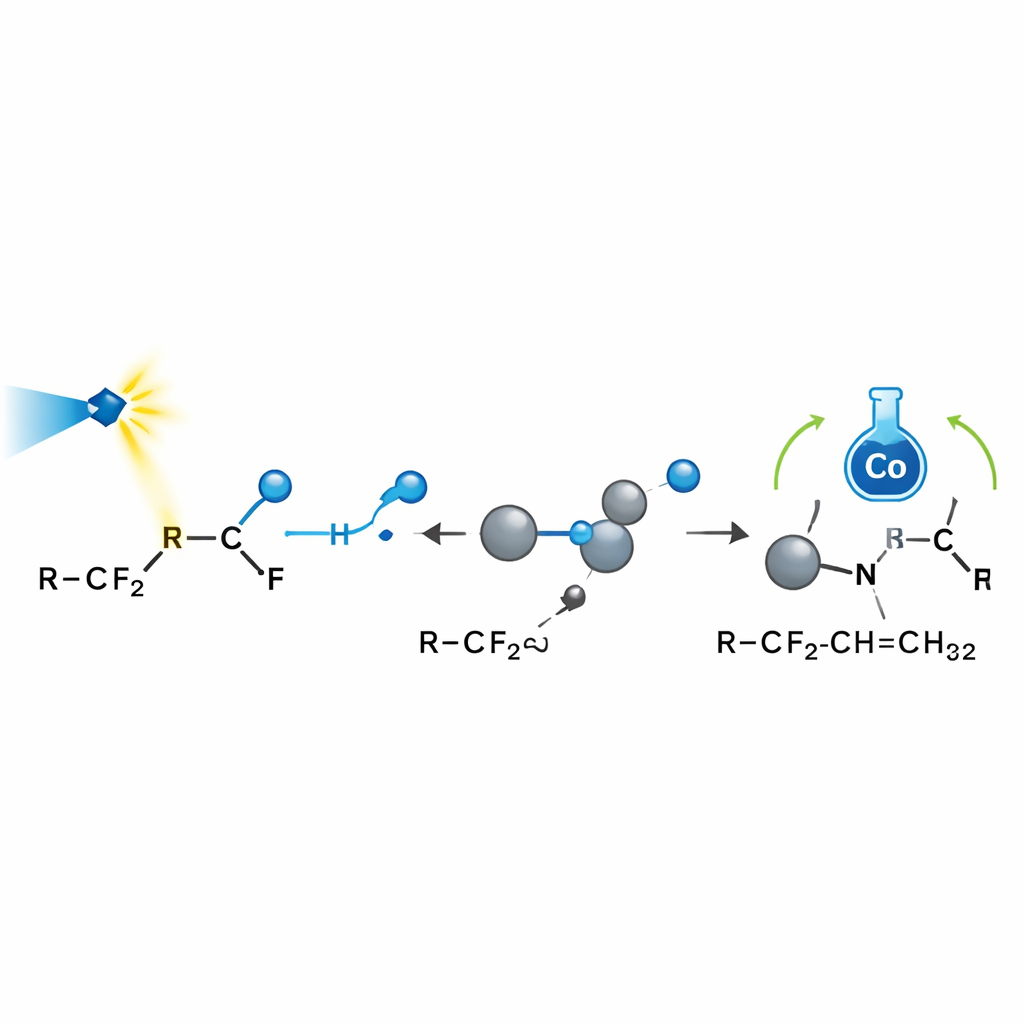
Explorer l'origine de la sélectivité
Pour comprendre pourquoi la réaction est si sélective, l'équipe a combiné des sondes expérimentales en laboratoire et de la modélisation informatique. Des « pièges » de radicaux et des expériences d'ouverture de cycle ont confirmé que les radicaux fluorés jouent un rôle central. Des mesures de résonance paramagnétique électronique ont montré que le dioxyde de carbone peut aussi être réduit en un anion réactif qui aide à activer certains amides fluorés. Des signaux de résonance magnétique nucléaire ont révélé que le réactif à base de bore capture effectivement le fluorure libre. Des calculs de chimie quantique ont cartographié le paysage énergétique de chaque étape, montrant qu'après l'addition du radical à un alcène, le cobalt préfère délivrer l'hydrogène à un site particulier parce qu'il rencontre moins d'encombrement stérique et bénéficie d'interactions attractives favorables. Cela explique à la fois pourquoi une seule position allylique réagit et pourquoi les produits favorisent fortement une seule configuration géométrique autour de la double liaison.
Ce que cette avancée signifie en pratique
En orchestrant la lumière, un catalyseur métallique et un additif fixant le fluorure, ce travail transforme certaines des liaisons carbone–fluor les plus robustes en connexions carbone–carbone sur mesure sans conditions rudes ni matériaux de départ élaborés. Concrètement, la méthode convertit des fragments fluorés extrêmement inertes en blocs de construction flexibles qui peuvent être attachés à des alcènes simples de manière prévisible. Cela ouvre une voie vers des échafaudages fluorés adaptés sur mesure pour la pharmacie, l'agrochimie et les matériaux avancés, tout en offrant de nouveaux outils pour repenser comment les substances fluorées persistantes pourraient être transformées plutôt que simplement subies.
Citation: Ren, D., Deng, S., Wang, Y. et al. Radical defluoroallylation of polyfluoroalkyl compounds with alkenes via synergistic photoredox/cobalt catalysis. Nat Commun 17, 2971 (2026). https://doi.org/10.1038/s41467-026-68840-3
Mots-clés: molécules fluorées, catalyse photoredox, catalyse au cobalt, réactions radicalaires, fonctionnalisation d'alcènes