Clear Sky Science · de
Radikale Defluoroallylierung von polyfluorierten Alkylverbindungen mit Alkenen durch synergetische Photoredox-/Kobalt‑Katalyse
Warum das Umwandeln hartnäckiger, fluorreicher Moleküle wichtig ist
Fluorreiche Verbindungen sind in der modernen Welt allgegenwärtig, von Arzneimitteln und Pflanzenschutzmitteln bis hin zu fleckenabweisenden Beschichtungen. Ihre Wirkung beruht auf Kohlenstoff–Fluor‑Bindungen, die zu den stärksten in der organischen Chemie gehören und diese Verbindungen im Körper und in der Umwelt besonders stabil machen. Dieselbe Härte macht sie jedoch sehr schwer gezielt zu verändern. Die vorliegende Studie stellt einen schonenden Weg vor, eine spezifische Kohlenstoff–Fluor‑Bindung in langen fluorierten Ketten aufzubrechen und durch eine nützliche Kohlenstoff–Kohlenstoff‑Bindung zu ersetzen, was Chemikern helfen könnte, bessere Wirkstoffe und Materialien aus diesen hartnäckigen Molekülen zu entwerfen.
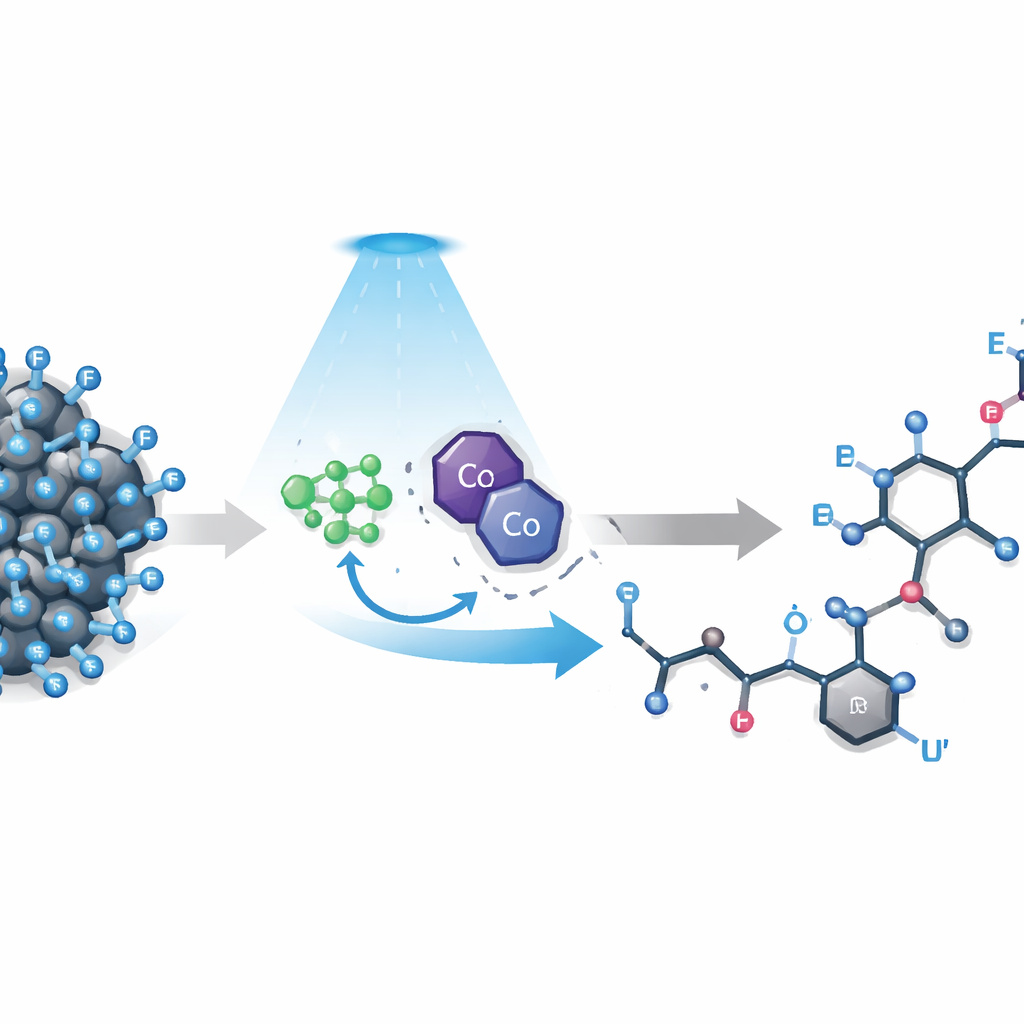
Die Herausforderung, die stärkste Bindung zu durchtrennen
Polyfluoralkylverbindungen enthalten Ketten, in denen viele Wasserstoffatome durch Fluor ersetzt wurden. Diese Ketten machen Moleküle wasserabweisend und chemisch widerstandsfähig — vorteilhaft für Technik und Medizin, aber problematisch, wenn Chemiker ihr Verhalten feinabstimmen wollen. Bestehende Methoden konzentrieren sich meist auf die einfachste fluorierte Gruppe, CF3, oder erfordern hochreaktive Zusatzstoffe und vorgefertigte Kopplungspartner. Längere Ketten, wie sie in realen Produkten häufig vorkommen, lassen sich noch schwerer kontrollieren: die starke Elektronegativität des Fluors destabilisiert reaktive Zwischenstufen, sperrige Fluoratome versperren den Zugang, und Elektronen können rückfließen und frühe Schritte rückgängig machen. Deshalb führen Versuche, genau eine Kohlenstoff–Fluor‑Bindung zu spalten, oft zu unübersichtlichen Gemischen oder dem Scheitern der Reaktion.
Ein zweifaches Katalysatorsystem, angetrieben durch Licht
Die Autoren entwickelten eine Strategie, die zwei kooperierende Katalysatoren und sichtbares Licht nutzt, um diese schwierigen Moleküle zu zähmen. Ein lichtempfindlicher Iridiumkomplex spendet zunächst ein Elektron an die polyfluorierte Verbindung, schwächt dadurch eine Kohlenstoff–Fluor‑Bindung gerade genug, damit sie bricht und ein kurzlebiges, fluoriertes Fragment — ein Radikal — freisetzt. Gleichzeitig lenkt ein kobaltbasierter Katalysator, der am Modell von Vitamin B12 orientiert ist, wohin schließlich ein Wasserstoffatom eingebracht wird. Ein einfaches Bor‑Reagenz wirkt als „Fluorid‑Schwamm“, fängt das ausgetriebene Fluor ein und verhindert, dass es wieder reagiert oder Elektronen zurückleitet. Unter milden Blau‑LED‑Bedingungen kanalisiert dieses Helfernetzwerk die Reaktion auf einen produktiven, hochselektiven Pfad.
Neue Bindungen zu einfachen Alkenen aufbauen
Einmal gebildet, addieren sich die fluorierten Radikale an einfache Alkene — kleine Moleküle mit einer Kohlenstoff‑Kohlenstoff‑Doppelbindung — und erzeugen in einem Schritt neue Kohlenstoff‑Kohlenstoff‑Bindungen. Der Kobalt‑Katalysator überträgt anschließend ein Wasserstoffatom auf eine spezifische Nachbarposition und verankert so ein „allylisches“ Fragment, während die nützliche Doppelbindung erhalten bleibt. In Dutzenden von Prüfungen funktionierte die Methode bei einer breiten Palette von Alkenen, einschließlich komplexer natürlicher Terpene, und bei vielen verschiedenen fluorreichen Ausgangsstoffen mit Kettenlängen von drei bis zehn Kohlenstoffen oder fluorierten Amidgruppen. Auffällig ist, dass selbst wenn Alkene mehrere nahezu identische allylische C–H‑Stellen enthalten, die Reaktion eine Position mit einer Selektivität von mehr als 20:1 bevorzugte und so das Isomerengemisch vermied, das diese Chemie sonst oft begleitet.
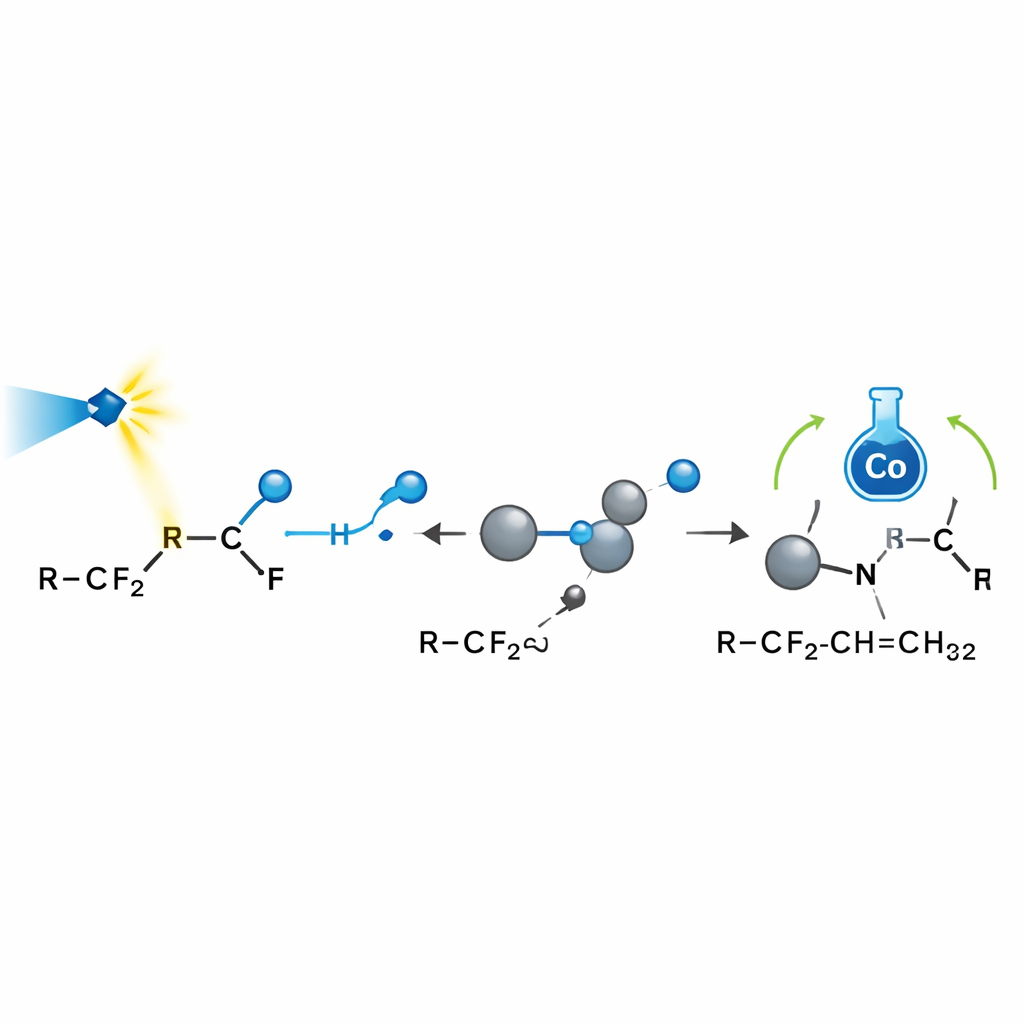
Untersuchung der Ursprünge der Selektivität
Um zu verstehen, warum die Reaktion so selektiv ist, kombinierten die Forschenden Laboruntersuchungen mit Computermodellen. Chemische „Radikalfallen“ und Ringöffnungs‑Experimente bestätigten, dass fluorierte Radikale Schlüsselakteure sind. Elektronenspinmessungen zeigten, dass Kohlendioxid ebenfalls zu einem reaktiven Anion reduziert werden kann, das bestimmte fluorierte Amide aktiviert. Kernspinresonanz‑Signale belegten, dass das Bor‑Reagenz tatsächlich freies Fluorid einfängt. Quantenchemische Berechnungen kartierten die Energielandschaft jedes Schrittes und zeigten, dass nachdem das Radikal an ein Alken addiert hat, Kobalt bevorzugt ein Wasserstoffatom an eine bestimmte Stelle liefert, weil dort weniger sterische Behinderung vorliegt und vorteilhafte anziehende Wechselwirkungen wirken. Das erklärt sowohl, warum nur eine allylische Position reagiert, als auch warum die Produkte eine starke Bevorzugung einer einzigen geometrischen Anordnung um die Doppelbindung zeigen.
Was dieser Fortschritt praktisch bedeutet
Indem Licht, ein Metallkatalysator und ein fluorideinbindendes Additiv orchestriert werden, wandelt diese Arbeit einige der härtesten Kohlenstoff–Fluor‑Bindungen in maßgeschneiderte Kohlenstoff–Kohlenstoff‑Verbindungen um, ohne raue Bedingungen oder aufwendige Ausgangsstoffe. Anschaulich verwandelt die Methode extrem inerte fluorierte Fragmente in flexible Bausteine, die vorhersehbar an einfache Alkene angehängt werden können. Das eröffnet einen Weg zu maßgeschneiderten fluorierten Gerüsten für Pharmazeutika, Pflanzenschutzmittel und fortschrittliche Materialien und bietet zugleich neue Werkzeuge, um darüber nachzudenken, wie persistente fluorierte Substanzen transformiert statt nur hingenommen werden können.
Zitation: Ren, D., Deng, S., Wang, Y. et al. Radical defluoroallylation of polyfluoroalkyl compounds with alkenes via synergistic photoredox/cobalt catalysis. Nat Commun 17, 2971 (2026). https://doi.org/10.1038/s41467-026-68840-3
Schlüsselwörter: fluorierte Moleküle, Photoredox‑Katalyse, Kobalt‑Katalyse, radikalische Reaktionen, Funktionalisierung von Alkenen