Clear Sky Science · en
Radical defluoroallylation of polyfluoroalkyl compounds with alkenes via synergistic photoredox/cobalt catalysis
Why changing tough fluorine-rich molecules matters
Fluorine-packed chemicals are everywhere in modern life, from medicines and crop protectants to stain-resistant coatings. Their power comes from carbon–fluorine bonds that are among the strongest in organic chemistry, making these compounds unusually stable in the body and the environment. That same toughness, however, makes them very hard to modify on purpose. This study introduces a gentle way to snip one specific carbon–fluorine bond in long fluorinated chains and swap it for a useful carbon–carbon link, potentially helping chemists design better drugs and materials from these stubborn molecules.
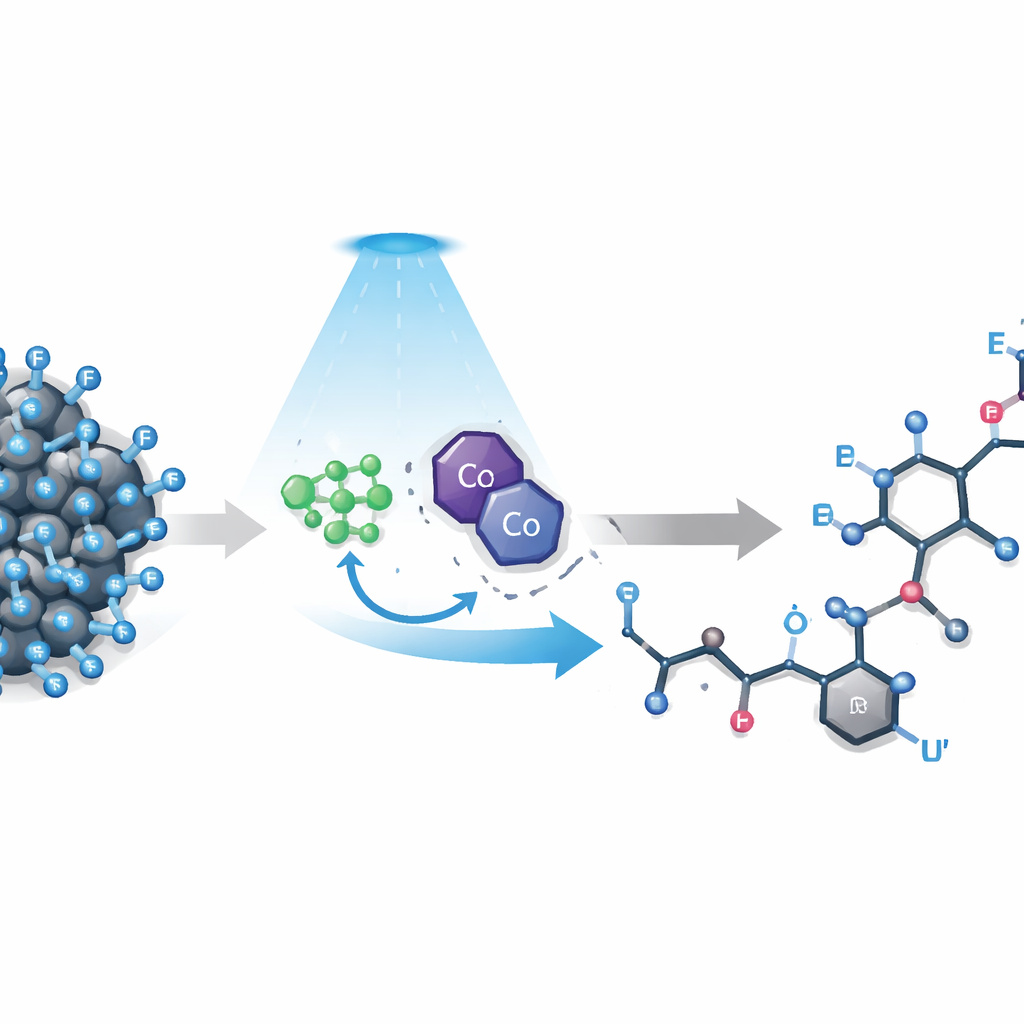
The challenge of cutting the strongest bond
Polyfluoroalkyl compounds contain chains where many hydrogens have been replaced by fluorine. These chains make molecules water‑repelling and chemically resilient, which is attractive for technology and medicine but problematic when chemists want to fine‑tune their behavior. Existing methods tend to focus on the simplest fluorinated group, CF3, or demand highly reactive additives and pre‑built coupling partners. Longer chains, which are common in real products, are even harder to control: the strong fluorine pull destabilizes reactive intermediates, bulky fluorine atoms block access, and electrons can flow backward to undo early steps. As a result, attempts to break one chosen carbon–fluorine bond often lead to messy mixtures or outright failure.
A two‑catalyst approach powered by light
The authors developed a strategy that uses two cooperating catalysts and visible light to tame these difficult molecules. A light‑sensitive iridium complex first donates a single electron to a polyfluoroalkyl compound, weakening one carbon–fluorine bond just enough for it to break and release a short‑lived fluorinated fragment known as a radical. At the same time, a cobalt catalyst modeled on vitamin B12 guides where a hydrogen atom will eventually be added. A simple boron reagent acts as a “fluoride sponge,” trapping the expelled fluorine and preventing it from recombining or shuttling electrons backward. Under mild blue‑LED illumination, this network of helpers channels the reaction along a productive, highly selective path.
Building new bonds with simple alkenes
Once formed, the fluorinated radicals add to everyday alkenes—small molecules with a carbon–carbon double bond—creating new carbon–carbon links in a single step. The cobalt catalyst then transfers a hydrogen atom to a specific neighboring position, locking in an “allyl” fragment while preserving the useful double bond. Across dozens of tests, the method worked on a wide range of alkenes, including complex natural terpenes, and on many different fluorine‑rich starting materials bearing chains from three to ten carbons long or fluorinated amide groups. Strikingly, even when alkenes contained several nearly identical allylic C–H sites, the reaction chose one position with better than 20‑to‑1 selectivity, avoiding the tangle of isomers that usually plagues such chemistry.
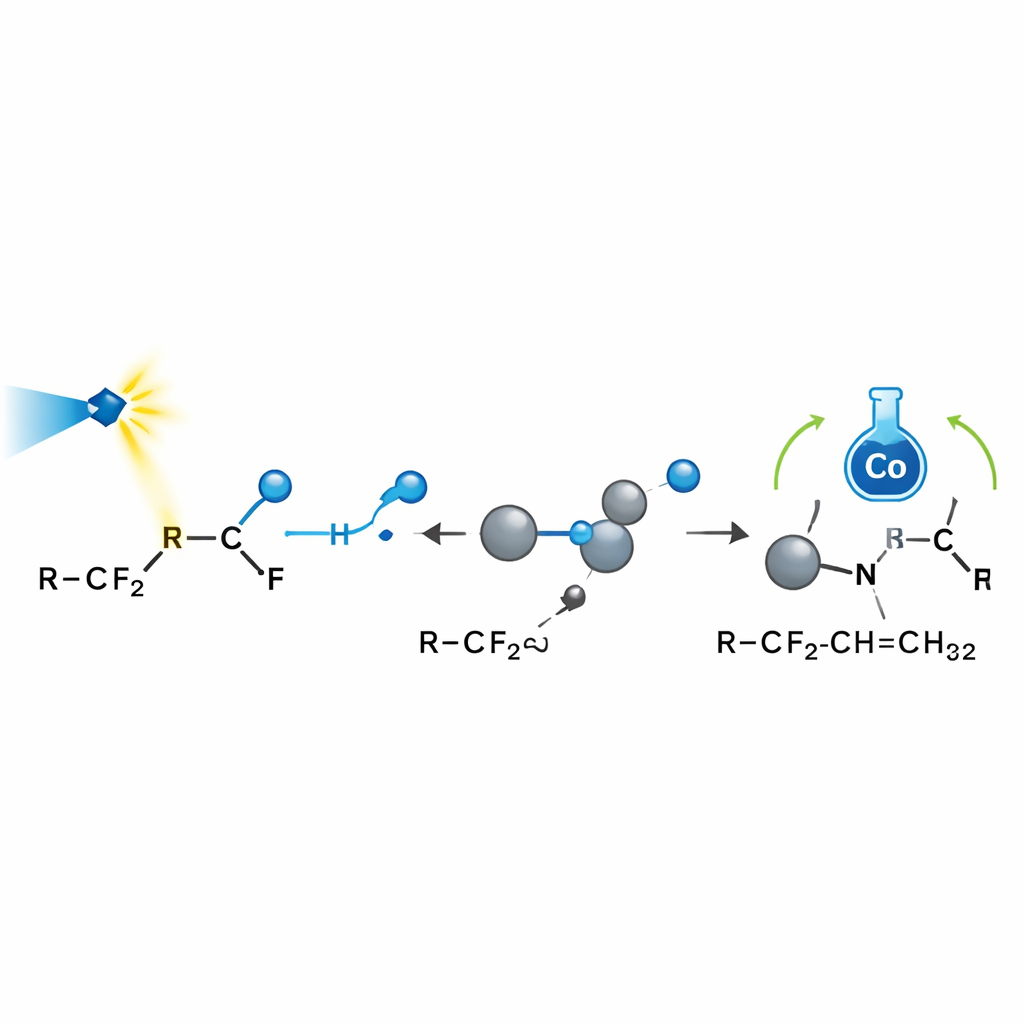
Probing how selectivity arises
To understand why the reaction is so selective, the team combined laboratory probes with computer modeling. Chemical “radical traps” and ring‑opening experiments confirmed that fluorinated radicals are key players. Electron‑spin measurements showed that carbon dioxide can also be reduced to a reactive anion that helps activate certain fluorinated amides. Nuclear magnetic resonance signals revealed that the boron reagent indeed captures free fluoride. Quantum‑chemical calculations mapped out the energy landscape of each step, showing that after the radical adds to an alkene, cobalt prefers to deliver hydrogen to one particular site because it faces less steric crowding and benefits from favorable attractive interactions. This explains both why only one allylic position reacts and why the products strongly favor a single geometric arrangement around the double bond.
What this advance means in practice
By orchestrating light, a metal catalyst, and a fluoride‑binding additive, this work converts some of the toughest carbon–fluorine bonds into designer carbon–carbon connections without harsh conditions or elaborate starting materials. In everyday terms, the method turns extremely inert fluorinated fragments into flexible building blocks that can be attached to simple alkenes in a predictable way. This opens a path to tailor‑made fluorinated scaffolds for pharmaceuticals, agrochemicals, and advanced materials, while also offering new tools to rethink how persistent fluorinated substances might be transformed rather than simply endured.
Citation: Ren, D., Deng, S., Wang, Y. et al. Radical defluoroallylation of polyfluoroalkyl compounds with alkenes via synergistic photoredox/cobalt catalysis. Nat Commun 17, 2971 (2026). https://doi.org/10.1038/s41467-026-68840-3
Keywords: fluorinated molecules, photoredox catalysis, cobalt catalysis, radical reactions, alkene functionalization