Clear Sky Science · fr
Libération d’ADN mitochondrial via VDAC1 dans les kératinocytes : un moteur clé de l’immunité innée et de la pathogenèse du vitiligo
Pourquoi cette histoire cutanée compte
Les taches blanches de la peau causées par le vitiligo peuvent être profondément éprouvantes ; pourtant les traitements actuels ne fonctionnent souvent que partiellement et les rechutes sont fréquentes. Cette étude explore un coupable surprenant à l’intérieur de cellules cutanées ordinaires : de minuscules centrales énergétiques appelées mitochondries. Les auteurs montrent comment un stress au sein de ces structures peut déclencher un signal d’alarme qui réveille le système immunitaire et favorise la perte de pigmentation. En retraçant cette chaîne d’événements pas à pas, ils désignent aussi une nouvelle piste thérapeutique qui ne cible pas directement le système immunitaire, mais la manière dont les cellules de la peau lancent d’abord l’appel à l’aide.
Les cellules de la peau comme émetteurs d’alarme actifs
Le vitiligo a longtemps été considéré comme une maladie où des cellules immunitaires attaquent par erreur les cellules productrices de pigment, les mélanocytes. Cependant, les cellules les plus externes de la peau — les kératinocytes — sont bien plus nombreuses que les mélanocytes et sont de plus en plus reconnues comme des sentinelles précoces de la maladie. Chez les personnes atteintes de vitiligo, la peau subit un fort « stress oxydatif », une tension chimique comparable à ce qui se produit sous un fort ensoleillement ou en cas de pollution. Les kératinocytes soumis à ce stress libèrent des messagers inflammatoires qui attirent des lymphocytes T cytotoxiques vers la peau. La question clé posée par les auteurs est : qu’est‑ce qui transforme exactement ce stress chimique en un puissant signal de danger qui active la machinerie antivirale et inflammatoire de l’organisme ?
Des centrales qui servent aussi de balises d’alerte
Les mitochondries portent leurs propres petits cercles de matériel génétique, l’ADN mitochondrial. Quand cet ADN s’échappe dans le corps principal de la cellule, le système immunitaire le traite souvent comme s’il provenait d’un virus ou d’une bactérie. Les chercheurs montrent que l’exposition des kératinocytes au peroxyde d’hydrogène — un agent classique de stress oxydatif — endommage les mitochondries sans tuer immédiatement les cellules. Au microscope, la structure mitochondriale devient déformée et des fragments d’ADN mitochondrial apparaissent à l’extérieur de l’organite tandis que la quantité totale d’ADN mitochondrial à l’intérieur de la cellule reste à peu près constante. Cet ADN fuitant active alors deux puissants systèmes de détection qui protègent normalement contre les infections, entraînant la production d’interférons (des protéines qui amplifient les réponses immunitaires) et de chimiokines (des signaux qui attirent les cellules immunitaires). 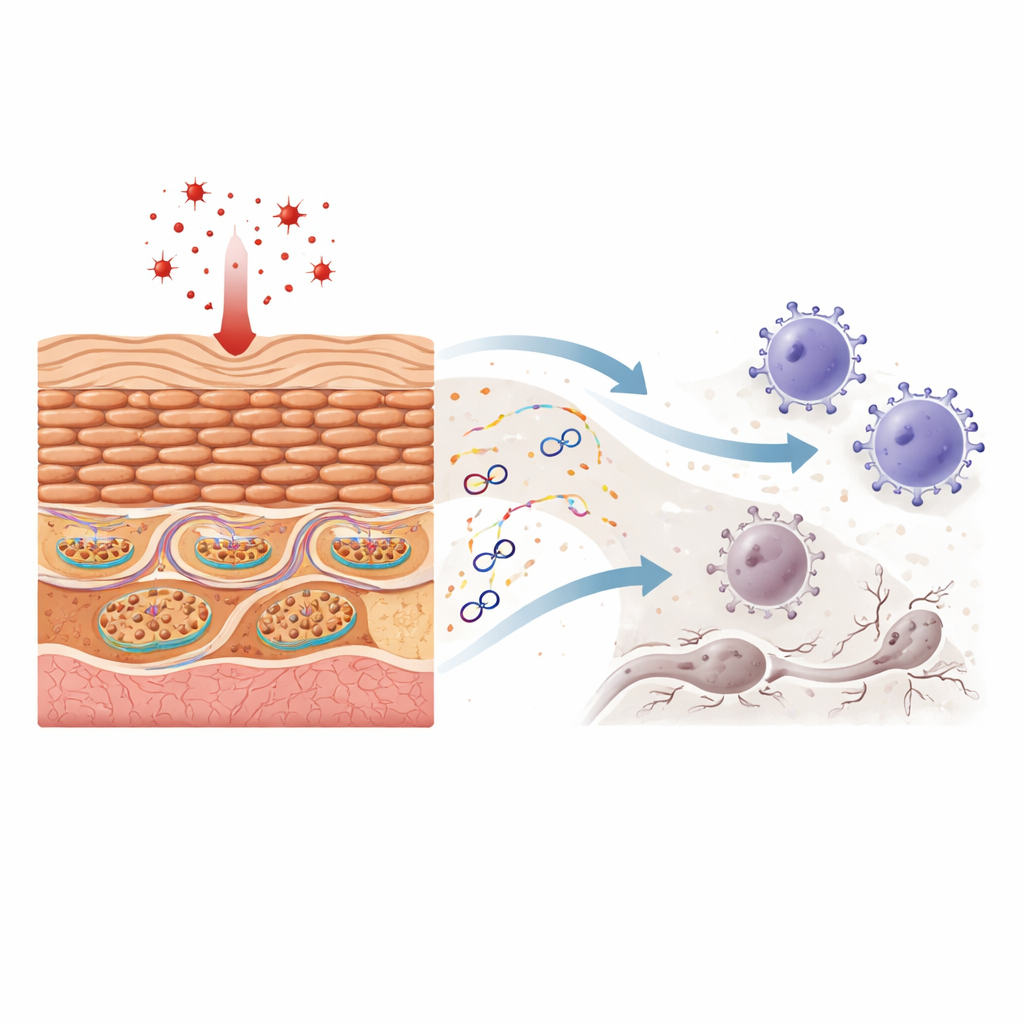
Comment l’ADN mitochondrial s’échappe de sa cage
Pour comprendre comment cet ADN sort, les auteurs se concentrent sur deux portes dans les membranes mitochondriales. L’une est le pore de transition de perméabilité dans la membrane interne, qui peut s’ouvrir sous l’effet du stress. L’autre est VDAC1, un canal de la membrane externe qui peut s’assembler en ouvertures plus larges. À l’aide de colorants pour cellules vivantes et de sondes moléculaires, ils montrent que le stress oxydatif ouvre d’abord le pore interne puis favorise l’agrégation de VDAC1, le tout sans déclencher les voies classiques du suicide cellulaire. Bloquer le pore interne empêche l’ADN de passer du noyau mitochondrial vers l’espace intermembranaire, tandis que bloquer VDAC1 l’empêche de franchir la frontière externe vers le cytosol. Les deux interventions réduisent fortement la quantité d’ADN mitochondrial dans le cytosol, confirmant une voie d’évasion étagée dépendant de VDAC1.
De l’alarme interne à la mort cellulaire enflammée
L’équipe a ensuite cherché à savoir si l’ADN mitochondrial errant suffit à activer les défenses des kératinocytes. Lorsqu’ils ont purifié cet ADN et l’ont introduit directement dans des kératinocytes, il a fortement activé le système cGAS–STING — un détecteur interne d’ADN — et un autre complexe nommé l’inflammasome. Ensemble, ces circuits ont entraîné des niveaux élevés d’interférons et de chimiokines et ont poussé les cellules vers la pyroptose, une forme explosive et inflammatoire de mort cellulaire qui perce la membrane cellulaire. Bloquer cGAS a atténué ces réponses et réduit la pyroptose, montrant que cette voie de détection de l’ADN se situe en amont d’une grande partie de la cascade inflammatoire. 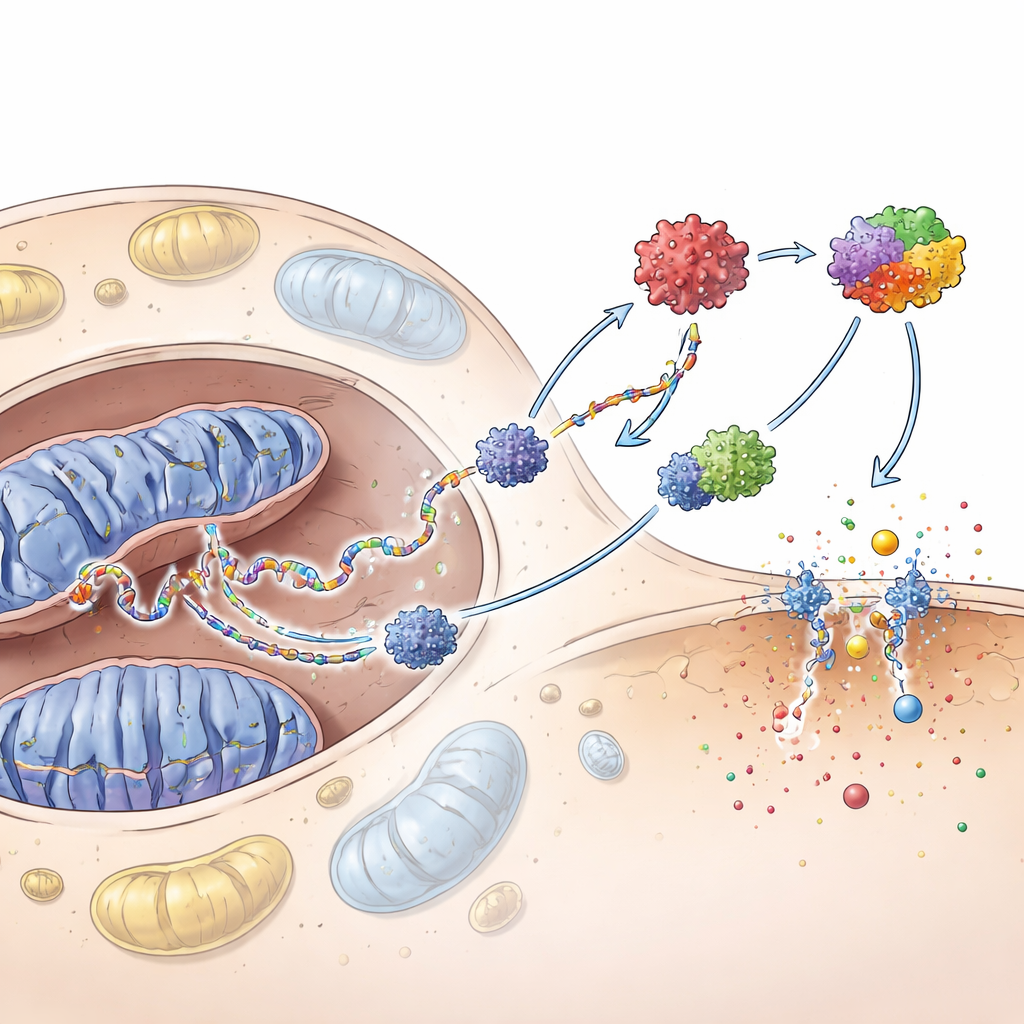
Fermer la porte pour apaiser la peau
Parce que VDAC1 agit comme une porte externe clé pour la fuite d’ADN, les chercheurs ont testé si fermer cette porte pouvait atténuer les signes de la maladie. En culture cellulaire, une petite molécule appelée VBIT‑4, qui empêche l’agrégation de VDAC1, a réduit la libération d’ADN mitochondrial et atténué l’activation à la fois du système cGAS–STING et de l’inflammasome, diminuant la production de chimiokines. L’inhibition de l’expression du gène VDAC1 a eu des effets similaires. Dans un modèle murin de vitiligo induit par application cutanée de peroxyde d’hydrogène, les animaux traités par VBIT‑4 ont développé moins de taches pâles, conservé davantage de mélanocytes et présenté moins de cellules CD8⁺ dans la peau. Leur peau montrait également des niveaux plus faibles des voies inflammatoires liées à l’ADN mitochondrial.
Une nouvelle façon d’intercepter précocement le vitiligo
Pour un non‑spécialiste, l’idée principale est que les cellules cutanées ordinaires ne restent pas passives pendant que le système immunitaire devient défaillant : elles contribuent au déclenchement du problème. Sous stress chimique, leurs mitochondries fuient des fragments d’ADN par des « portes » VDAC1, et cet ADN fuitant agit comme une fausse alarme d’infection. Cette alarme réveille des programmes antiviraux et inflammatoires puissants, attire des cellules immunitaires agressives et favorise des formes destructrices de mort cellulaire, préparant le terrain à la perte de pigment. En ciblant la porte VDAC1, des médicaments comme VBIT‑4 pourraient un jour étouffer cette alarme à sa source, offrant un moyen de ralentir ou prévenir le vitiligo avant que des dégâts irréversibles aux cellules pigmentaires ne surviennent.
Citation: Lv, J., Xu, W., Jiang, P. et al. Mitochondrial DNA release via VDAC1 in keratinocytes: a key driver of innate immunity and vitiligo pathogenesis. Cell Death Dis 17, 318 (2026). https://doi.org/10.1038/s41419-026-08585-5
Mots-clés: vitiligo, ADN mitochondrial, kératinocytes, immunité innée, VDAC1