Clear Sky Science · en
Non-necroptotic MLKL function damages mitochondria and promotes hematopoietic stem cell aging
Why Our Blood’s Lifelong Factory Wears Out
Deep inside our bones, a small pool of stem cells quietly builds every blood and immune cell we need throughout life. With age, this factory falters: infections linger, anemia becomes more common, and blood cancers such as myelodysplastic syndromes appear more often. This study uncovers a hidden molecular culprit—an unexpected behavior of a protein called MLKL—that slowly injures the stem cells’ powerhouses and helps drive the aging of our blood system.
Stress Signals That Don’t Quite Kill
Hematopoietic stem cells, the rare “master” cells in bone marrow, are unusually resistant to dying, even when bombarded by inflammation or chemotherapy. The authors focused on a molecular pathway normally used to trigger a dramatic form of cell death called necroptosis, driven by two proteins, RIPK3 and MLKL. Using engineered mice carrying a fluorescent sensor that lights up when MLKL is activated, they found that a range of common stresses—inflammatory molecules that mimic viral infection, bacterial components, and a chemotherapy drug that forces stem cells to divide—reliably switched on MLKL in stem cells and their closest progeny, but not in more mature blood precursors. Surprisingly, despite this activation, the stem cells did not die in large numbers. Instead, those with highly active MLKL showed poorer ability to engraft and make lymphoid cells when transplanted into new mice, hinting that MLKL was quietly damaging function rather than triggering outright death.
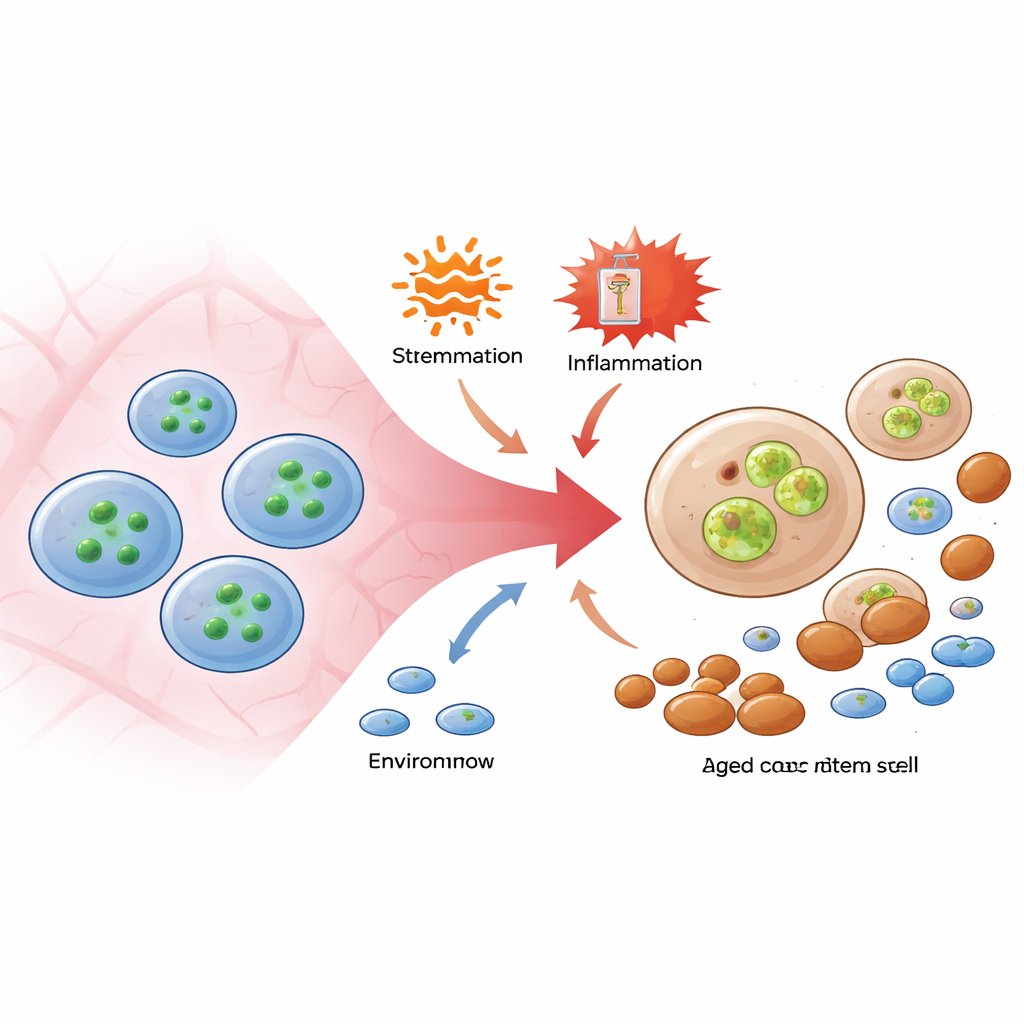
A Damage Pathway Hidden Inside Surviving Cells
To tease apart death from dysfunction, the researchers used mice lacking MLKL, or carrying a version of the protein that cannot be turned on by RIPK3. When these animals were challenged with inflammatory stimuli or repeated chemotherapy, their stem cells survived about as well as those in normal mice, and the total number of stem cells in bone marrow stayed similar. However, the usual age-like shifts—expansion of stem cells biased toward producing myeloid cells and loss of balanced stem cells capable of making both lymphoid and myeloid lineages—were markedly blunted when MLKL activity was removed. Even more striking, when MLKL-deficient stem cells were forced to face an oncogenic stress that ordinarily models pre‑leukemic disease, the animals were protected from severe bone marrow failure. These experiments showed that MLKL can undermine stem-cell quality from within, even when it is prevented from executing its classical cell-death program.
From Wear and Tear to True Aging
The team then asked whether this non‑lethal MLKL activity helps explain normal aging. In older mice, the MLKL sensor glowed more brightly in stem cells, revealing that this pathway becomes chronically engaged with age. Deleting MLKL did not stop stem cells from accumulating over time, but it did lessen classic hallmarks of aging: blood production became less skewed toward myeloid cells, early lymphoid progenitors were better preserved, and DNA damage markers in stem cells were reduced. Transplant tests confirmed that aged stem cells lacking MLKL retained stronger regenerative power and more balanced production of T and B lymphocytes. Importantly, the surrounding bone-marrow environment in aged mice showed similar levels of inflammatory molecules whether MLKL was present or not, and broad surveys of gene activity and chromatin structure in stem cells revealed little difference between normal old cells and those missing MLKL. That suggests MLKL acts mainly through a physical, rather than genetic, mode of damage.
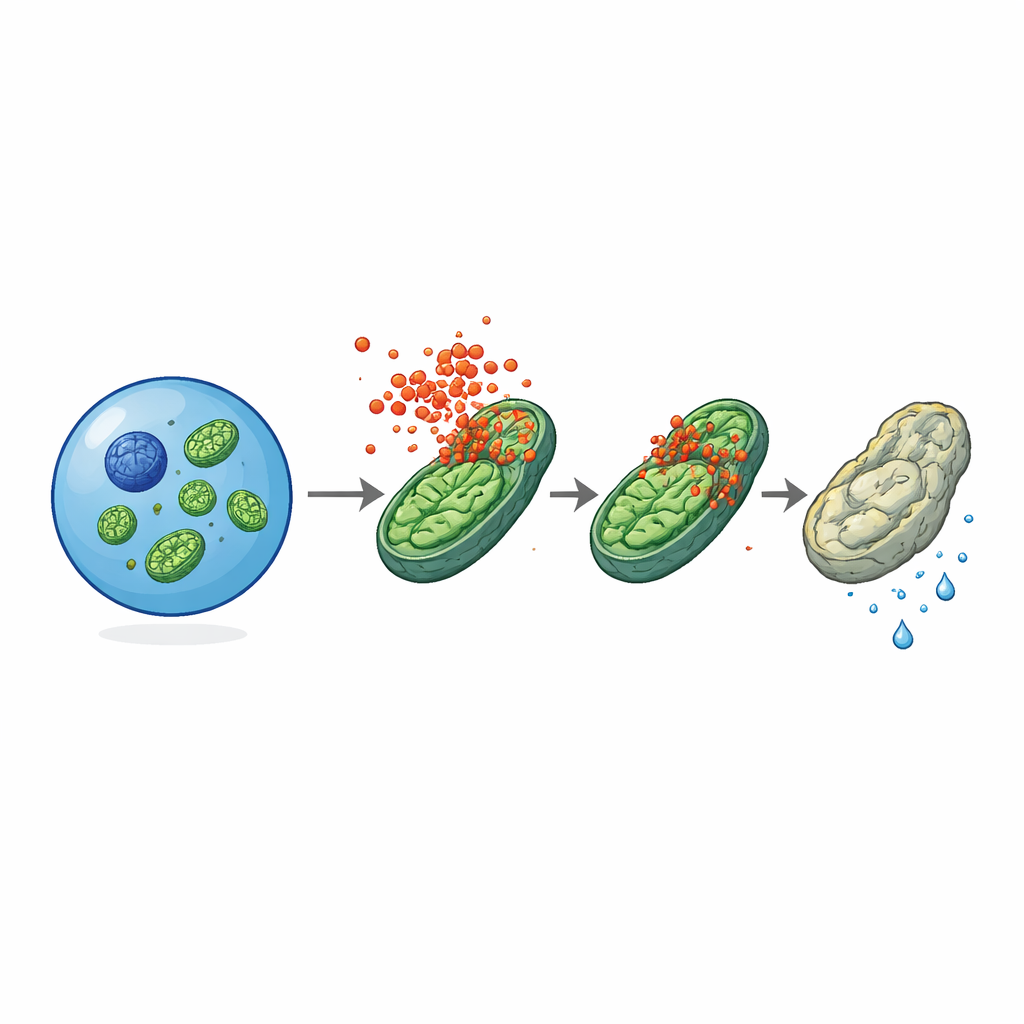
Powerhouses Under Attack
Electron microscopy gave a crucial clue: mitochondria—the tiny organelles that generate energy—looked swollen and misshapen in old stem cells from normal mice, but appeared far healthier in age‑matched cells lacking MLKL. The active, phosphorylated form of MLKL was found clustering on mitochondrial membranes, especially in aged or inflamed stem cells. Functional tests showed that old stem cells without MLKL had better mitochondrial membrane potential, produced more ATP, and maintained higher glycolytic activity than their normal counterparts, suggesting that MLKL gradually pushes their metabolism away from a youthful, glycolysis‑leaning state toward a more stressed, oxygen‑hungry mode. In isolated mitochondria exposed to purified MLKL fragments, the protein directly reduced membrane potential, indicating it can poke or destabilize mitochondrial membranes even outside the full cellular context.
What This Means for Healthy Aging
Taken together, the work reveals that MLKL, long known as an executioner of catastrophic cell death, also moonlights as a slow saboteur of stem-cell mitochondria. Repeated bouts of inflammation, replication stress, and oncogenic insults appear to converge on this pathway, allowing stem cells to survive immediate crises but at the cost of cumulative damage that blunts their regenerative capacity and tilts blood production toward riskier patterns. While MLKL and its partners can be beneficial for fighting infections and curbing cancer in certain contexts, carefully tuning this pathway—or shielding mitochondria from its non‑lethal attacks—may one day help preserve a younger, more resilient blood system as we age.
Citation: Yamada, Y., Yang, J., Saiki-Tsuchiya, A. et al. Non-necroptotic MLKL function damages mitochondria and promotes hematopoietic stem cell aging. Nat Commun 17, 2798 (2026). https://doi.org/10.1038/s41467-026-71060-4
Keywords: hematopoietic stem cell aging, mitochondrial dysfunction, MLKL, inflammation and chemotherapy stress, blood stem cell metabolism