Clear Sky Science · en
Rafoxanide disrupts mitochondrial homeostasis through VDAC1 modulation in colorectal cancer cells
Why this study matters
Colorectal cancer is one of the most common and deadly cancers worldwide. While many treatments attack DNA or block growth signals, cancer cells often survive by rewiring how they make and use energy. This study explores how an old veterinary drug, rafoxanide, can be repurposed to strike at cancer’s “power stations” – the mitochondria – in a precise way that pushes tumor cells toward self-destruction while largely sparing healthy tissue.
The cell’s power stations under pressure
Mitochondria are tiny structures inside cells that generate most of the cell’s energy and help decide whether a cell lives or dies. Cancer cells, including those in colorectal tumors, depend heavily on well-functioning mitochondria to fuel rapid growth and spread. The researchers focused on a gatekeeper protein called VDAC1, which sits in the outer skin of mitochondria and controls the flow of energy-carrying molecules in and out. When VDAC1 changes shape and clusters together, it can form large openings that allow death-promoting factors to escape into the cell.
An old parasite drug with a new target
Rafoxanide was originally designed to kill parasitic worms by disturbing their energy production. Earlier work showed it could slow colorectal tumor growth and trigger stress in another compartment of the cell, the endoplasmic reticulum, but how it affected mitochondria in human cancer cells was unclear. In this study, the team treated colorectal cancer cell lines with rafoxanide and measured oxygen use, energy production, and the electrical charge across the mitochondrial membrane. Within minutes, the drug sharply reduced mitochondrial breathing and began to weaken the membrane’s charge, yet this early effect was reversible when the drug was washed away, suggesting a targeted disturbance rather than blunt poisoning.
From reversible stress to fatal damage
When the treatment was extended to hours, the picture changed. Layering gene activity data, protein profiles, and metabolite measurements, the researchers found broad and lasting disruptions to mitochondrial components and pathways that help shuttle fuel, build new molecules, and balance cellular chemistry. Cancer cells failed to mount a successful adaptation, their mitochondrial membrane gradually lost its charge, and a key protein called cytochrome c leaked from mitochondria into the surrounding fluid – a classic early step in programmed cell death. Shortly afterward, the cells began to die, confirming that prolonged mitochondrial stress had crossed a point of no return.
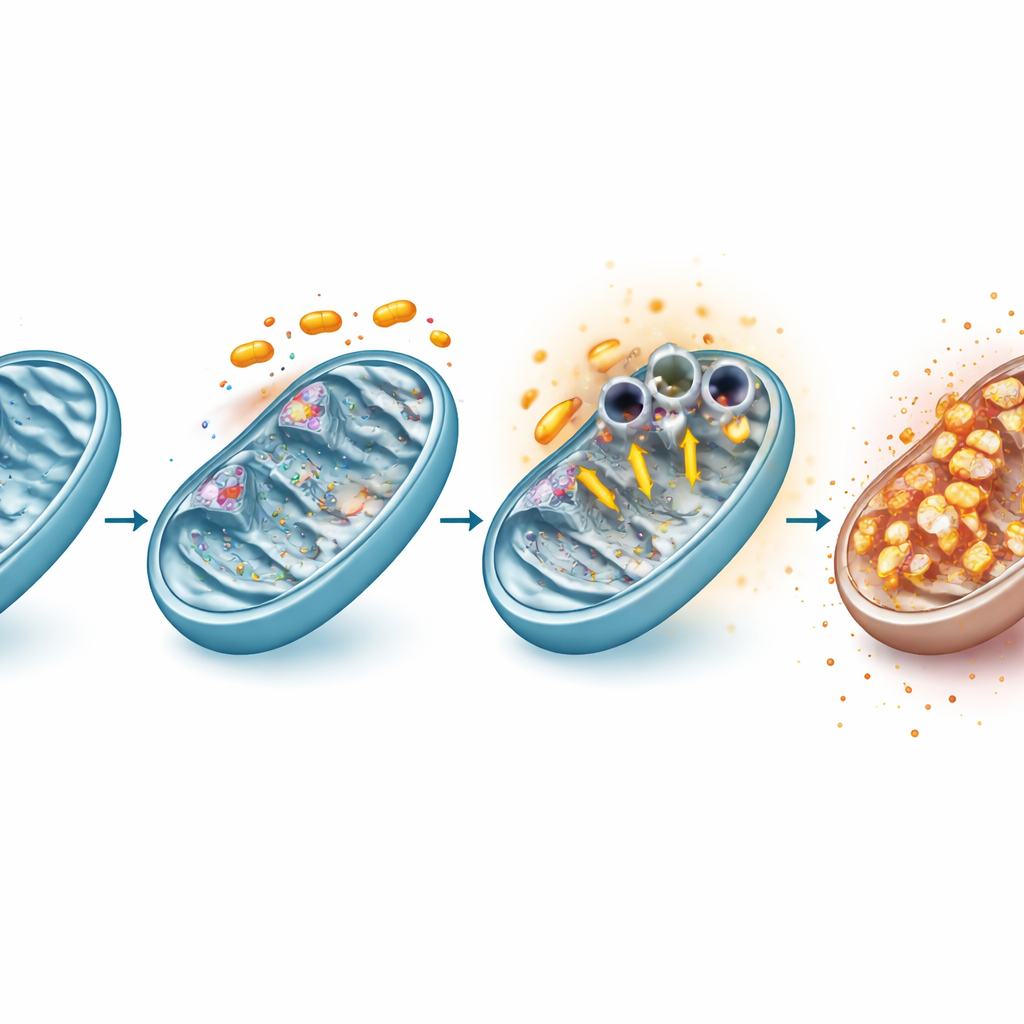
How the mitochondrial gate is forced open
Digging deeper, the team showed that rafoxanide directly dampens two major protein assemblies in the mitochondrial energy chain, known as complexes I and III. This interference rapidly altered the cell’s redox balance, leading to a burst of reactive oxygen species – chemically reactive forms of oxygen – even as mitochondria themselves produced less of a specific byproduct called superoxide because their internal charge was collapsing. This redox disturbance drove VDAC1 molecules in the outer mitochondrial membrane to cluster into larger structures, effectively opening wide channels. Blocking reactive oxygen species with an antioxidant, or pharmacologically inhibiting VDAC1 clustering, reduced both the opening of these channels and the loss of mitochondrial membrane charge, tying oxidative stress, VDAC1 behavior, and mitochondrial failure into a single chain of events.
Evidence beyond the dish
To test whether these effects occur in more realistic settings, the researchers turned to mice with chemically induced colorectal tumors and to samples taken directly from patients. In mice, rafoxanide treatment led to fewer and smaller tumors and reduced signs of cell proliferation. Detailed protein analysis of tumor tissue revealed broad shifts in mitochondrial proteins: those involved in burning fuel and running the energy chain tended to decline, while proteins linked to breaking down damaged components and burning fats rose, hinting at cells struggling to cope with energy stress. Similar mitochondrial protein changes appeared in patient-derived tumor explants and three-dimensional intestinal organoids exposed to rafoxanide, indicating that this mitochondrial disruption is consistent across cell cultures, animal models, and human tumor tissue.
What this means for future cancer treatment
Overall, the study shows that rafoxanide does not simply “pull the plug” on energy production; instead, it nudges colorectal cancer cell mitochondria into a prolonged state of dysfunction they cannot escape. By blocking key energy chain complexes, disturbing the balance of reactive molecules, and driving the VDAC1 gate to open, the drug sets off a controlled sequence that ends in mitochondrial failure and cell death. Normal colon cells, which are less dependent on stressed mitochondria and have more reserve capacity, appear far less affected. These findings raise the possibility that carefully tuned mitochondrial stress – rather than outright destruction – could be a powerful way to selectively weaken cancer cells, and they support further exploration of rafoxanide-like compounds as future therapies for colorectal cancer.
Citation: Tomassini, L., Pacifico, T., Serra, M.A. et al. Rafoxanide disrupts mitochondrial homeostasis through VDAC1 modulation in colorectal cancer cells. Cell Death Discov. 12, 142 (2026). https://doi.org/10.1038/s41420-026-02986-3
Keywords: colorectal cancer, mitochondria, VDAC1, reactive oxygen species, drug repurposing