Clear Sky Science · zh
MSCMF-DTB:一种用于药物–靶点结合预测的多尺度跨模态融合框架
为什么更智能的药物匹配很重要
找出哪些药物会与体内哪些蛋白结合,就像在解一个由数百万个碎片组成的巨大三维拼图。实验室里逐一测试所有可能的药物–蛋白对既缓慢又昂贵,因此研究人员正转向人工智能来缩小搜索范围。本文提出了MSCMF-DTB,一种新的深度学习系统,旨在同时预测药物是否会与蛋白结合以及结合强度。通过在单一模型中整合多种分子信息,它旨在加速药物发现、指导药物重定位并提高虚拟筛选的可靠性。
从多角度看待药物与蛋白
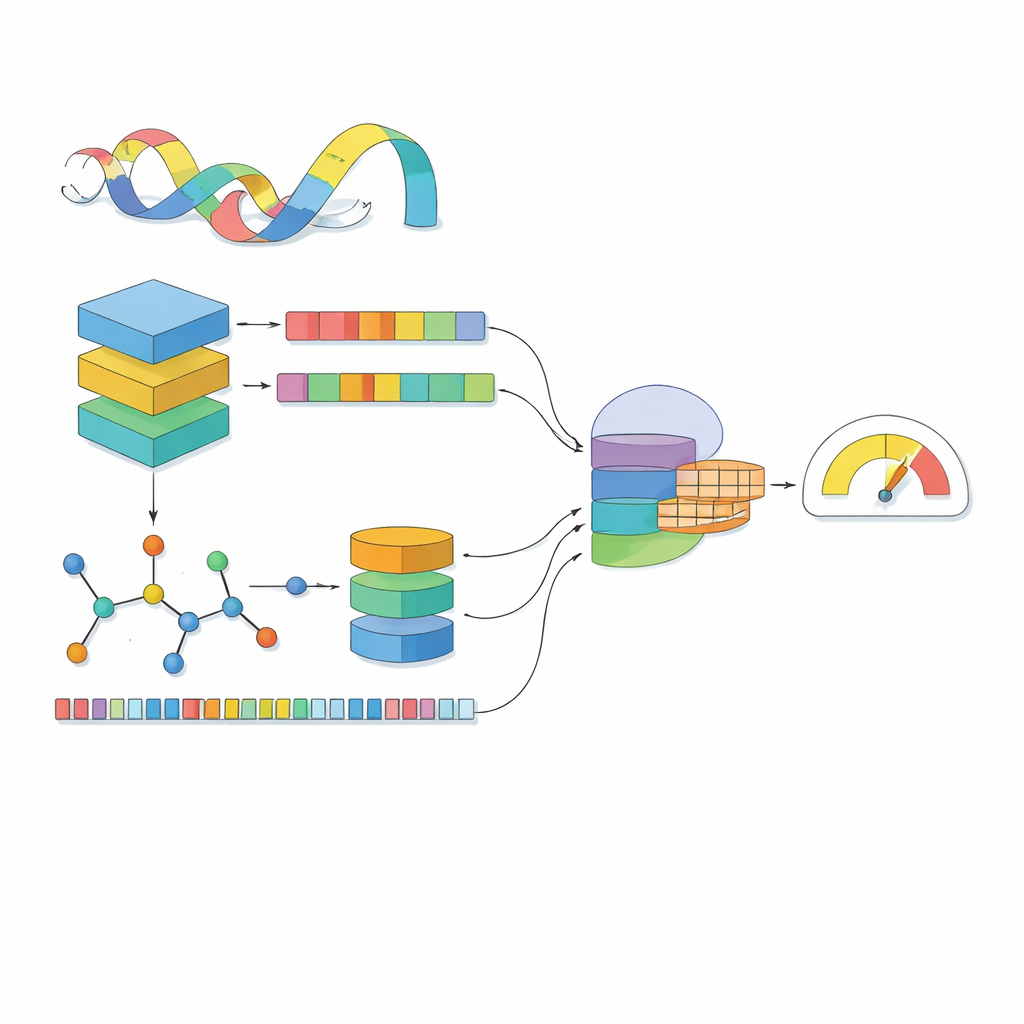
大多数计算模型将药物视为一串符号,将蛋白视为一串字母。MSCMF-DTB更进一步:它既将每种药物视为由原子和化学键构成的网络,也视为重复出现的化学片段的模式;同时将蛋白视为借助数百万条天然蛋白学习到的上下文信息而丰富的序列。对于药物,模型构建了以原子为节点、以化学键为边的图结构,并将该图输入深层网络,从小范围邻域逐步学习到整个分子的模式。并行地,第二通道捕捉特定原子类型和片段出现频率,提供对同一药物的互补性汇总视角。对于蛋白,强大的类语言模型首先将每个氨基酸转化为带有上下文信息的向量,然后用几种不同尺寸的一维滤波器扫描这些向量,以捕捉局部基序和序列上的远程模式。
教模型注意相互影响
单独观察药物和蛋白还不够;关键在于它们相遇时如何相互影响。MSCMF-DTB使用跨注意力机制,使药物表征可以有选择地“查看”蛋白的不同区域,反之亦然。这让模型能够突出哪些原子和蛋白片段最可能相互作用。除此之外,一个专门的张量网络学习药物与蛋白特征的高阶非线性组合,超越了简单特征混合。图分支、指纹分支、蛋白分支和交互分支的输出随后被合并并传入最终预测模块,该模块既可执行二分类的结合/不结合判断,也可输出连续的亲和力评分。
将系统付诸测试
作者在该领域广泛使用的一系列基准集合上评估了MSCMF-DTB。对于是否结合的判断,他们在五个数据集上进行了测试,这些数据集覆盖人类、模式线虫物种、一类信号受体以及诸如BioSNAP和DrugBank之类的大型药物–蛋白目录。对于结合强度,他们使用了DAVIS和KIBA这两个针对激酶的标准数据集,包含测量的亲和力值。在小型和大型集合中,该模型表现出强劲且一致的性能,经常匹配或超越领先方法。例如,在大型DrugBank数据集上,与先前最优模型相比,其ROC曲线下面积最多提升了3.2%,召回率提升了6.1%,表明它在数据规模和多样性面前能找到更多真实相互作用而不崩溃。
深入观察并进行现实检验
为了确保模型并非黑箱,研究人员检查了其注意力机制在具有已知药物结合位点的真实蛋白结构上聚焦的位置。当他们将高注意力区域映射到三维蛋白模型上时,许多被高亮的残基与实验确认的结合区域重叠,这表明系统正在学习具有生物学意义的信号。他们还对癌症相关靶点AKT1进行了苛刻的“冷启动”实验,完全将该蛋白从训练集中排除。在需要对数千个候选化合物进行排序时,模型将若干已知的AKT1抑制剂推到前列,表明它能推广到先前未见的靶点并支持虚拟筛选和重定位场景。
这对未来药物意味着什么
简单来说,MSCMF-DTB是一个在药物与蛋白之间进行多视角匹配的工具。通过结合精细的原子图、片段统计和丰富的蛋白序列上下文,并显式建模这些元素之间的相互作用,它比许多现有方法提供了更准确和更稳定的预测。虽然它不能替代实验室试验,但可以大大缩小有前景候选物的范围并提示可能形成重要接触的部位。与基于物理的模拟和实验验证相结合,像MSCMF-DTB这样的框架有望使从分子到药物的漫长且昂贵的过程变得更快、更便宜、更具信息性。
引用: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
关键词: 药物–靶点相互作用, 深度学习在药物发现中的应用, 虚拟筛选, 结合亲和力预测, 跨模态融合