Clear Sky Science · fr
MSCMF-DTB : un cadre de fusion croisée multi‑échelle pour la prédiction de la liaison médicament–cible
Pourquoi des appariements de médicaments plus intelligents comptent
Déterminer quels médicaments se lient à quelles protéines dans notre organisme revient à résoudre un gigantesque puzzle 3D composé de millions de pièces. Tester chaque paire médicament–protéine en laboratoire est lent et coûteux : les chercheurs se tournent donc vers l’intelligence artificielle pour restreindre les recherches. Cet article présente MSCMF-DTB, un nouveau système d’apprentissage profond conçu pour prédire à la fois si un médicament se liera à une protéine et avec quelle intensité. En combinant plusieurs types d’informations moléculaires dans un seul modèle, il vise à accélérer la découverte de médicaments, orienter le repositionnement de composés et rendre le criblage virtuel plus fiable.
Analyser les médicaments et les protéines sous de nombreux angles
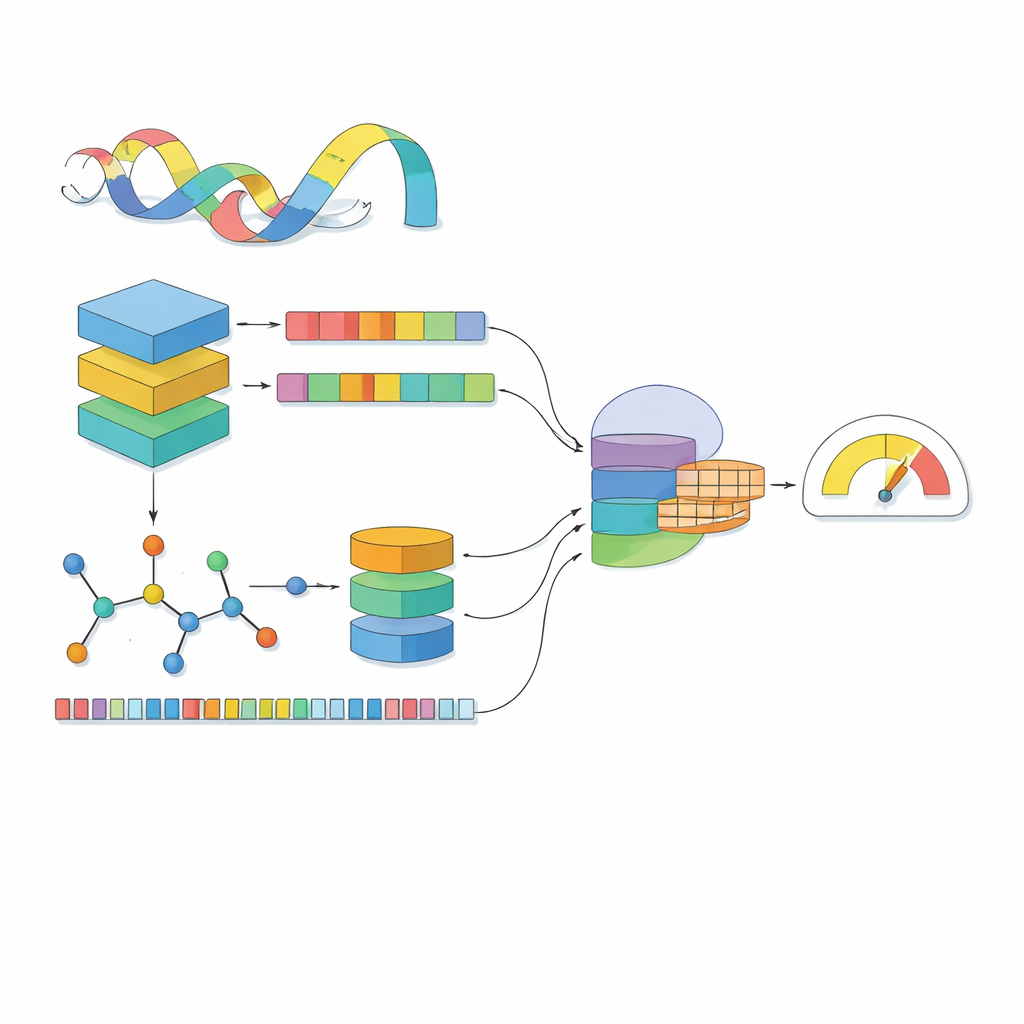
La plupart des modèles informatiques considèrent un médicament comme une chaîne de symboles et une protéine comme une suite de lettres. MSCMF-DTB va plus loin : il traite chaque médicament à la fois comme un réseau d’atomes et de liaisons et comme un motif de fragments chimiques récurrents, tandis que la protéine est représentée comme une séquence enrichie d’un contexte appris à partir de millions de protéines naturelles. Pour le médicament, le modèle construit un graphe où les atomes sont des nœuds et les liaisons des arêtes, puis fait passer ce graphe dans un réseau profond qui apprend progressivement des motifs des voisinages restreints jusqu’à l’ensemble de la molécule. En parallèle, un second canal capte la fréquence d’apparition de certains types d’atomes et de fragments, offrant une vue complémentaire de synthèse sur le même médicament. Pour la protéine, un puissant modèle de type « langage » transforme d’abord chaque acide aminé en un vecteur sensible au contexte, lequel est ensuite parcouru par plusieurs filtres unidimensionnels de tailles différentes pour repérer des motifs locaux et des schémas à plus longue portée le long de la séquence.
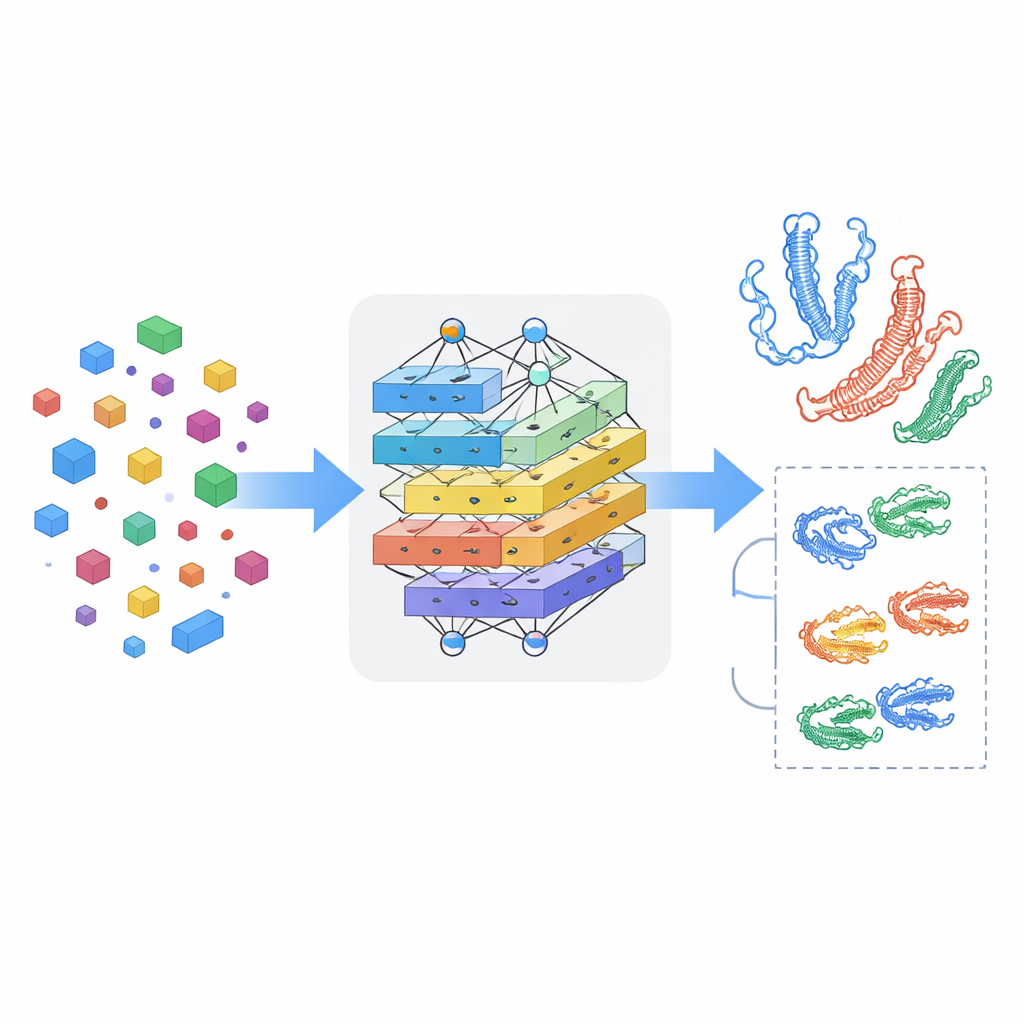
Apprendre au modèle à repérer les interactions croisées
Voir les médicaments et les protéines séparément ne suffit pas : l’essentiel tient à la manière dont ils s’influencent lorsqu’ils se rencontrent. MSCMF-DTB utilise un mécanisme d’attention croisée qui permet à la représentation du médicament de « regarder » sélectivement différentes régions de la protéine, et inversement. Cela permet au modèle de mettre en évidence quels atomes et quels segments protéiques sont les plus susceptibles d’interagir. De plus, un réseau tensoriel spécialisé apprend des combinaisons d’ordre supérieur et non linéaires des caractéristiques du médicament et de la protéine, dépassant le simple mélange de caractéristiques. Les sorties de la branche graphe, de la branche empreinte, de la branche protéine et de la branche interaction sont ensuite fusionnées et transmises à un module de prédiction final capable d’effectuer soit une classification binaire de liaison (oui/non), soit un score d’affinité continu.
Mettre le système à l’épreuve
Les auteurs ont évalué MSCMF-DTB sur un large ensemble de collections de référence largement utilisées dans le domaine. Pour les décisions oui/non de liaison, ils ont testé cinq jeux de données couvrant des échantillons humains, une espèce de ver modèle, une famille de récepteurs de signalisation et de grands catalogues médicament–protéine tels que BioSNAP et DrugBank. Pour la force de liaison, ils ont utilisé DAVIS et KIBA, deux jeux de données standard centrés sur les kinases contenant des valeurs d’affinité mesurées. Sur des collections petites et grandes, le modèle a montré des performances fortes et cohérentes, égalant souvent voire dépassant les méthodes de pointe. Sur le grand jeu de données DrugBank, par exemple, il a amélioré l’aire sous la courbe ROC jusqu’à 3,2 % et le rappel de 6,1 % par rapport au meilleur modèle antérieur, ce qui signifie qu’il a trouvé plus d’interactions réelles sans être perturbé par la taille et la diversité des données.
Regarder à l’intérieur et tester un scénario réaliste
Pour éviter que le modèle ne soit une simple boîte noire, les chercheurs ont examiné où son mécanisme d’attention se focalisait sur des structures protéiques réelles avec des sites de liaison connus. Lorsqu’ils ont superposé les régions de forte attention sur des modèles protéiques tridimensionnels, de nombreux résidus mis en évidence coïncidaient avec des régions de liaison confirmées expérimentalement, suggérant que le système apprend des signaux biologiquement pertinents. Ils ont aussi réalisé une expérience exigeante de « cold-start » sur la cible liée au cancer AKT1, en excluant complètement cette protéine de l’entraînement. En demandant au modèle de classer des milliers de composés candidats, plusieurs inhibiteurs connus d’AKT1 ont été propulsés parmi les meilleures prédictions, montrant qu’il peut généraliser à des cibles inédites et soutenir des scénarios de criblage virtuel et de repositionnement.
Ce que cela signifie pour les médicaments de demain
En termes simples, MSCMF-DTB est un entremetteur multi‑vue entre médicaments et protéines. En combinant des graphes atomiques détaillés, des statistiques de fragments et un contexte riche des séquences protéiques, et en modélisant explicitement la manière dont ces éléments communiquent entre eux, il offre des prédictions plus précises et plus stables que de nombreuses approches existantes. S’il ne remplace pas les expériences de laboratoire, il peut considérablement réduire la liste de candidats prometteurs et indiquer où des contacts importants sont susceptibles de se former. Associés à des simulations basées sur la physique et à des validations expérimentales, des cadres comme MSCMF-DTB pourraient contribuer à rendre le long et coûteux parcours de la molécule au médicament plus rapide, moins onéreux et mieux informé.
Citation: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Mots-clés: interaction médicament–cible, apprentissage profond en découverte de médicaments, criblage virtuel, prédiction de l’affinité de liaison, fusion cross-modale