Clear Sky Science · pl
MSCMF-DTB: wieloskalowe ramy fuzji międzymodalnej do przewidywania wiązania leku z celem
Dlaczego inteligentniejsze dopasowanie leków ma znaczenie
Ustalenie, które leki wiążą się z jakimi białkami w naszym organizmie, przypomina układanie ogromnej trójwymiarowej układanki z milionami elementów. Testowanie każdego możliwego parowania lek–białko w laboratorium jest powolne i kosztowne, dlatego badacze sięgają po sztuczną inteligencję, aby zawęzić poszukiwania. W pracy tej przedstawiono MSCMF-DTB, nowy system oparty na głębokim uczeniu zaprojektowany do przewidywania zarówno faktu wiązania leku z białkiem, jak i siły tego wiązania. Łącząc kilka rodzajów informacji molekularnej w jednym modelu, ma on przyspieszyć odkrywanie leków, wspierać repurposing i zwiększyć niezawodność screeningu wirtualnego.
Wielokątne spojrzenie na leki i białka
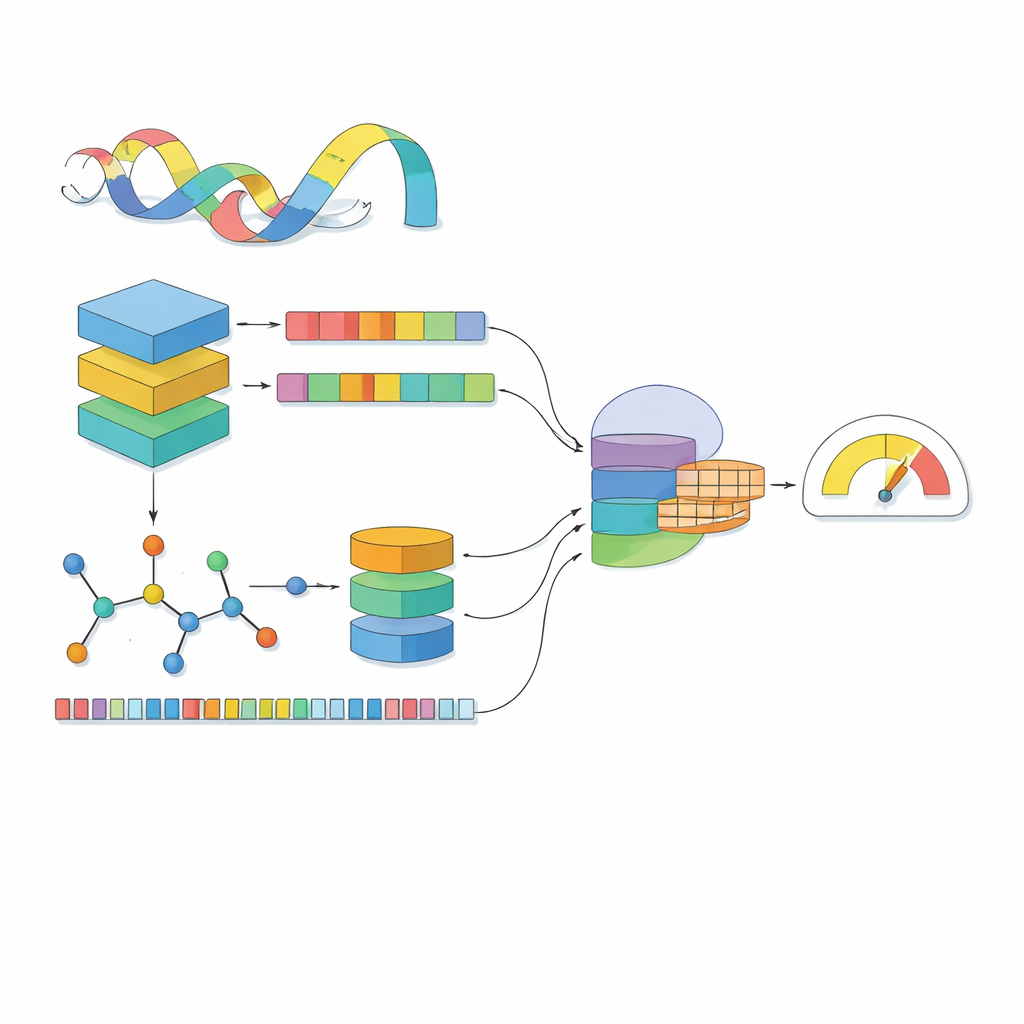
Większość modeli komputerowych traktuje lek jak linię symboli, a białko jak ciąg liter. MSCMF-DTB idzie dalej, widząc lek zarówno jako sieć atomów i wiązań, jak i wzór powtarzających się fragmentów chemicznych, natomiast białko jako sekwencję wzbogaconą kontekstem wyuczonym na milionach naturalnych białek. Dla leku model buduje graf, w którym atomy są węzłami, a wiązania krawędziami, a następnie przekazuje ten graf przez głęboką sieć, która stopniowo uczy się wzorców od małych sąsiedztw po całą cząsteczkę. Równolegle drugi kanał rejestruje częstość występowania określonych typów atomów i fragmentów, oferując uzupełniający, syntetyczny obraz tej samej cząsteczki. Dla białka potężny model typu „językowego” najpierw przekształca każdy aminokwas w wektor zależny od kontekstu, który następnie jest skanowany przez kilka jednowymiarowych filtrów o różnej szerokości, aby wychwycić lokalne motywy i dłuższe wzorce wzdłuż sekwencji.
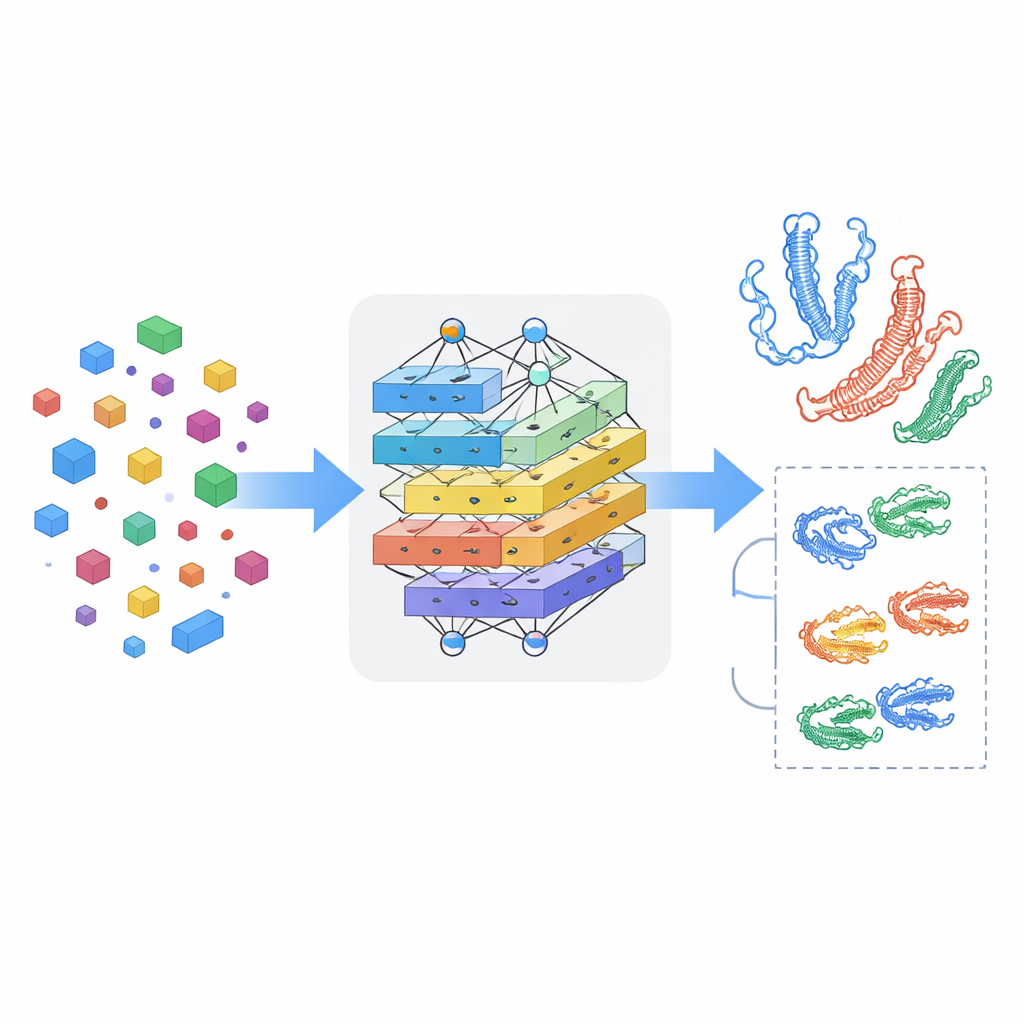
Nauczanie modelu rozpoznawania wzajemnych wpływów
Przyglądanie się lekom i białkom osobno nie wystarcza; kluczowe jest to, jak oddziałują ze sobą w zetknięciu. MSCMF-DTB wykorzystuje mechanizm cross-attention, dzięki któremu reprezentacja leku może selektywnie „patrzeć” na różne regiony białka i odwrotnie. Pozwala to modelowi wyróżnić atomy i segmenty białka, które mają największe szanse na interakcję. Ponadto wyspecjalizowana sieć tensorowa uczy się wyższych, nieliniowych kombinacji cech leku i białka, wykraczając poza proste łączenie cech. Wyjścia z gałęzi grafowej, gałęzi fingerprint, gałęzi białkowej oraz gałęzi interakcji są następnie scalane i przekazywane do końcowego modułu predykcyjnego, który może wykonywać zarówno klasyfikację tak/nie w kwestii wiązania, jak i ciągłe prognozowanie powinowactwa.
Testowanie systemu
Autorzy ocenili MSCMF-DTB na szerokim zestawie benchmarków powszechnie stosowanych w dziedzinie. Dla decyzji tak/nie dotyczących wiązania przetestowano go na pięciu zbiorach obejmujących białka ludzkie, modelowego nicienia, rodzinę receptorów sygnałowych oraz duże katalogi lek–białko, takie jak BioSNAP i DrugBank. Dla siły wiązania użyto DAVIS i KIBA, dwóch standardowych zbiorów ukierunkowanych na kinazy zawierających zmierzone wartości powinowactwa. W zbiorach małych i dużych model wykazał mocne i spójne wyniki, często dorównując lub przewyższając czołowe metody. Na dużym zbiorze DrugBank, na przykład, poprawił pole pod krzywą ROC o do 3,2% i recall o 6,1% w porównaniu z poprzednio najlepszym modelem, co oznaczało znalezienie większej liczby prawdziwych interakcji bez załamania przy skali i różnorodności danych.
Zajrzenie do wnętrza oraz realistyczny test
Aby upewnić się, że model nie jest jedynie czarną skrzynką, badacze zbadali, na które obszary struktury białek o znanych miejscach wiążących leki koncentrował się mechanizm uwagi. Po odwzorowaniu obszarów o wysokiej uwadze na trójwymiarowe modele białek wiele z wyróżnionych reszt pokrywało się z eksperymentalnie potwierdzonymi regionami wiążącymi, co sugeruje, że system uczy się biologicznie istotnych sygnałów. Przeprowadzili też wymagający eksperyment „cold-start” na celu związanym z rakiem AKT1, całkowicie wykluczając to białko z treningu. Po poproszeniu modelu o wybranie spośród tysięcy kandydatów, kilka znanych inhibitorów AKT1 znalazło się w czołowych prognozach, co pokazuje, że model potrafi uogólniać na wcześniej niewidziane cele i wspierać scenariusze screeningu wirtualnego oraz repurposing.
Co to oznacza dla przyszłych leków
Mówiąc prosto, MSCMF-DTB to wielopodglądowy swat łączący leki z białkami. Łącząc szczegółowe grafy atomowe, statystyki fragmentów oraz bogaty kontekst sekwencji białkowej i jawnie modelując, jak te elementy ze sobą rozmawiają, oferuje bardziej precyzyjne i stabilne przewidywania niż wiele istniejących podejść. Choć nie zastąpi eksperymentów laboratoryjnych, może znacznie zawęzić listę obiecujących kandydatów i zasugerować, gdzie mogą powstawać istotne kontakty. W połączeniu z symulacjami opartymi na fizyce i weryfikacją eksperymentalną, ramy takie jak MSCMF-DTB mogą przyspieszyć, obniżyć koszty i uczynić bardziej świadomą długą i kosztowną drogę od cząsteczki do leku.
Cytowanie: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Słowa kluczowe: interakcja lek–cel, uczenie głębokie w odkrywaniu leków, screening wirtualny, przewidywanie powinowactwa wiązania, fuzja międzymodalna