Clear Sky Science · en
MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction
Why Smarter Drug Matching Matters
Finding which medicines stick to which proteins inside our bodies is like solving a gigantic 3D puzzle with millions of pieces. Testing every possible drug–protein pair in the lab is slow and expensive, so researchers are turning to artificial intelligence to narrow the search. This paper introduces MSCMF-DTB, a new deep-learning system designed to predict both whether a drug will bind to a protein and how strongly it will bind. By combining several types of molecular information in one model, it aims to speed up drug discovery, guide drug repurposing, and make virtual screening more reliable.
Looking at Drugs and Proteins from Many Angles
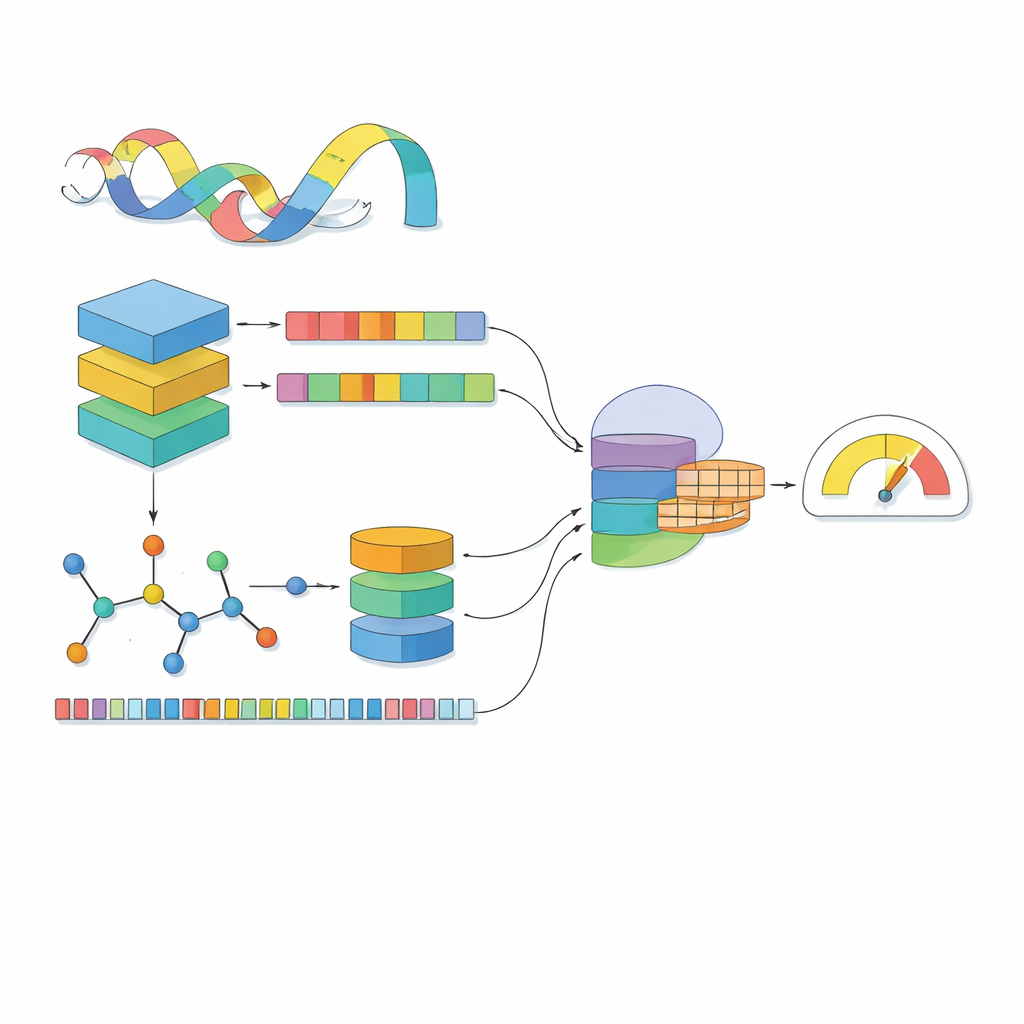
Most computer models see a drug as a line of symbols and a protein as a string of letters. MSCMF-DTB goes further by viewing each drug as both a network of atoms and bonds and a pattern of recurring chemical fragments, while treating the protein as a sequence enriched with context learned from millions of natural proteins. For the drug, the model builds a graph where atoms are points and bonds are links, then passes this graph through a deep network that gradually learns patterns from small neighborhoods up to the whole molecule. In parallel, a second channel captures how often certain atom types and fragments appear, offering a complementary, summary-style view of the same drug. For the protein, a powerful language-like model first turns each amino acid into a context-aware vector, which is then scanned by several one‑dimensional filters of different sizes to pick up local motifs and longer-range patterns along the sequence.
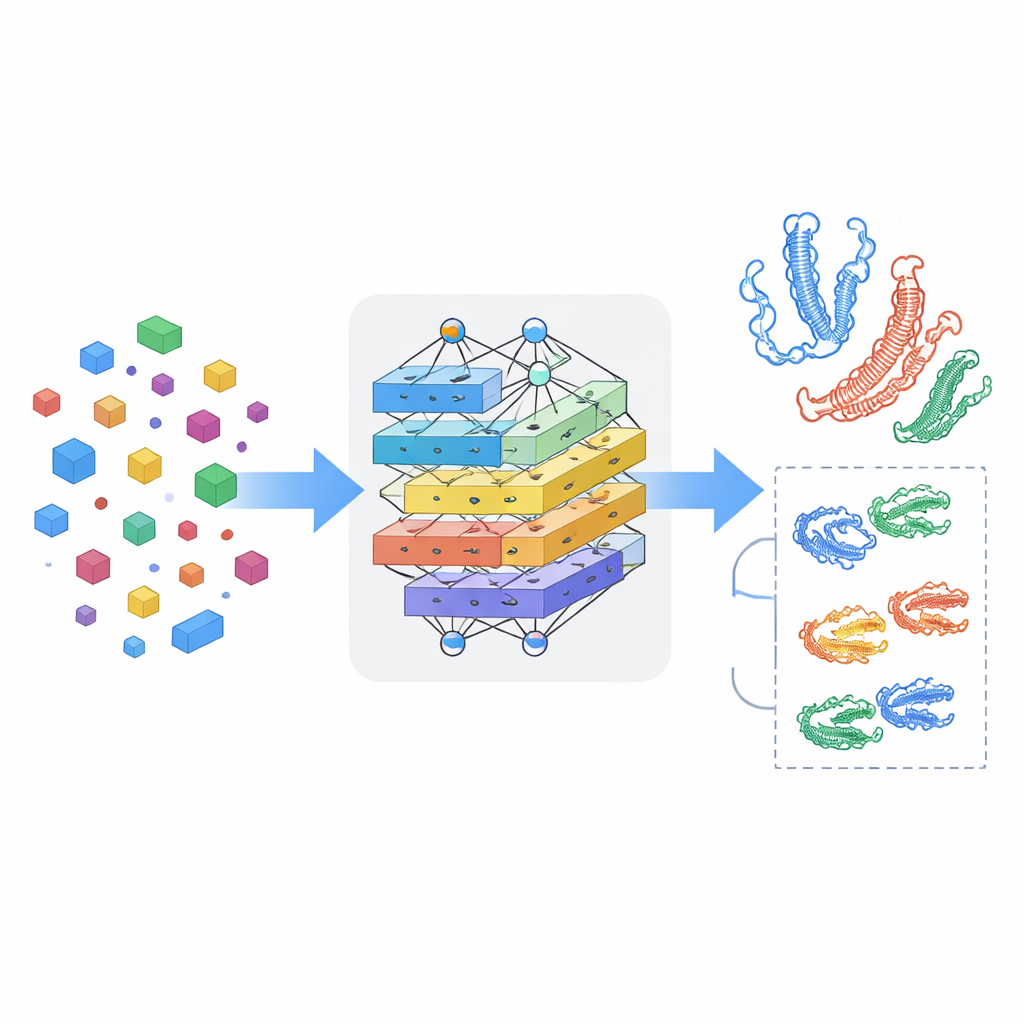
Teaching the Model to Notice Cross-Talk
Seeing drugs and proteins separately is not enough; the key is how they influence each other when they meet. MSCMF-DTB uses a cross-attention mechanism so that the drug’s representation can selectively “look at” different regions of the protein, and vice versa. This lets the model highlight which atoms and which protein segments are most likely to interact. On top of that, a specialized tensor network learns higher-order, non‑linear combinations of drug and protein features, going beyond simple feature mixing. The outputs of the graph branch, fingerprint branch, protein branch, and interaction branch are then merged and passed into a final prediction module that can perform either yes/no binding classification or continuous affinity scoring.
Putting the System to the Test
The authors evaluated MSCMF-DTB on a broad set of benchmark collections that are widely used in the field. For binding yes/no decisions, they tested on five datasets covering humans, a model worm species, a family of signaling receptors, and large drug–protein catalogs such as BioSNAP and DrugBank. For binding strength, they used DAVIS and KIBA, two standard kinase-focused datasets containing measured affinity values. Across small and large collections, the model showed strong and consistent performance, often matching or surpassing leading methods. On the large DrugBank dataset, for example, it improved the area under the ROC curve by up to 3.2% and recall by 6.1% compared with the previous best model, meaning it found more true interactions without collapsing under the size and diversity of the data.
Peeking Inside and Trying a Realistic Test
To make sure the model is not just a black box, the researchers examined where its attention mechanism focused on real protein structures with known drug-binding sites. When they mapped the high-attention regions onto three-dimensional protein models, many of the highlighted residues overlapped with experimentally confirmed binding regions, suggesting that the system is learning biologically meaningful signals. They also ran a demanding “cold-start” experiment on the cancer-related target AKT1, completely excluding this protein from training. When asked to rank thousands of candidate compounds, the model pushed several known AKT1 inhibitors into the top predictions, showing that it can generalize to previously unseen targets and support virtual screening and repurposing scenarios.
What This Means for Future Medicines
In simple terms, MSCMF-DTB is a multi-view matchmaker between drugs and proteins. By combining detailed atomic graphs, fragment statistics, and rich protein sequence context, and by explicitly modeling how these pieces talk to each other, it offers more accurate and stable predictions than many existing approaches. While it does not replace laboratory experiments, it can greatly narrow down the list of promising candidates and suggest where important contacts may form. Coupled with physics-based simulations and experimental validation, frameworks like MSCMF-DTB could help make the long and costly journey from molecule to medicine faster, cheaper, and more informed.
Citation: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Keywords: drug–target interaction, deep learning in drug discovery, virtual screening, binding affinity prediction, cross-modal fusion