Clear Sky Science · pt
MSCMF-DTB: uma estrutura de fusão cross-modal em múltiplas escalas para predição de ligação droga–alvo
Por que um Pareamento de Fármacos mais Inteligente Importa
Descobrir quais medicamentos se ligam a quais proteínas no nosso organismo é como resolver um enorme quebra-cabeça 3D com milhões de peças. Testar cada par possível droga–proteína em laboratório é lento e caro, por isso pesquisadores recorrem à inteligência artificial para reduzir a busca. Este artigo apresenta o MSCMF-DTB, um novo sistema de aprendizado profundo projetado para prever tanto se uma droga vai se ligar a uma proteína quanto quão fortemente ela se ligará. Ao combinar vários tipos de informação molecular em um único modelo, o objetivo é acelerar a descoberta de fármacos, orientar o reposicionamento de medicamentos e tornar a triagem virtual mais confiável.
Olhando para Drogas e Proteínas por Muitas Perspectivas
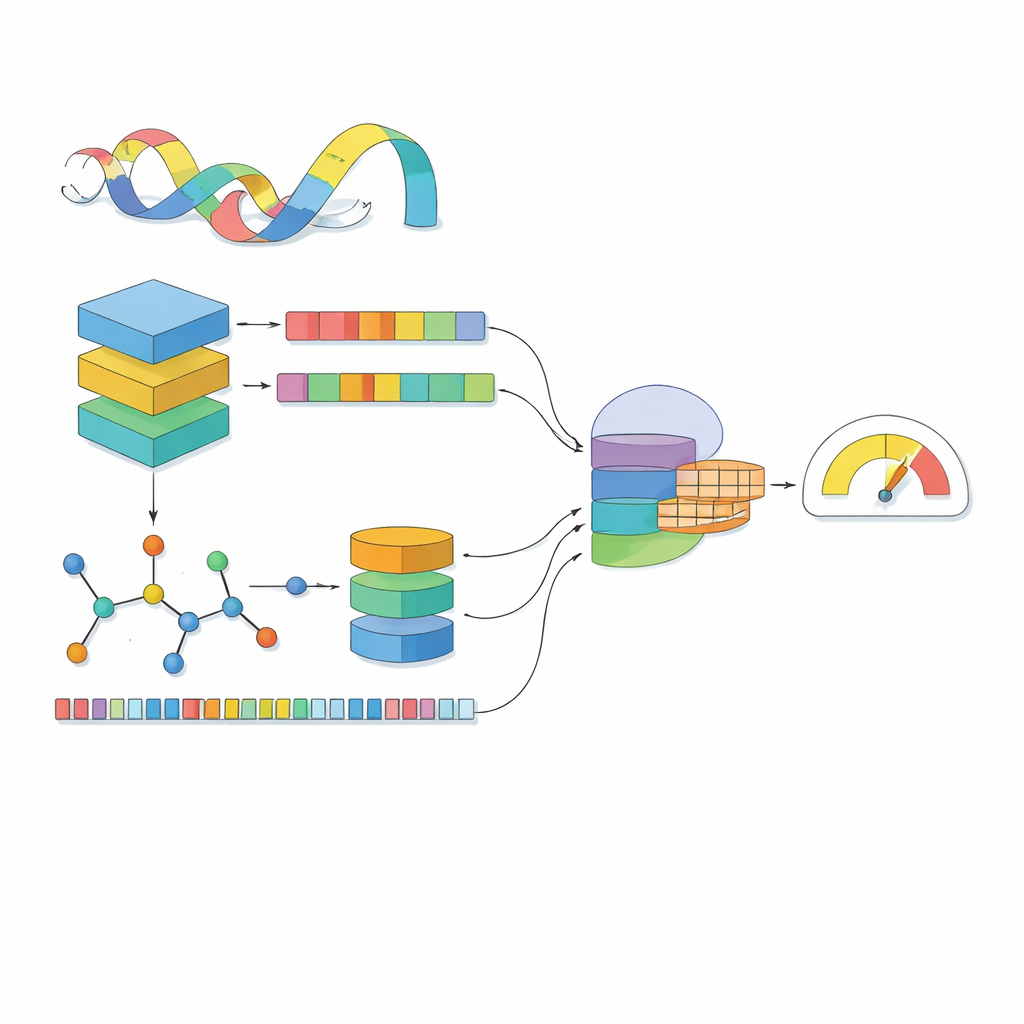
A maioria dos modelos computacionais vê uma droga como uma linha de símbolos e uma proteína como uma sequência de letras. O MSCMF-DTB vai além, tratando cada droga tanto como uma rede de átomos e ligações quanto como um padrão de fragmentos químicos recorrentes, enquanto considera a proteína como uma sequência enriquecida com contexto aprendido a partir de milhões de proteínas naturais. Para a droga, o modelo constrói um grafo onde átomos são nós e ligações são arestas, e então passa esse grafo por uma rede profunda que aprende gradualmente padrões desde vizinhanças pequenas até a molécula inteira. Em paralelo, um segundo canal captura a frequência de certos tipos de átomos e fragmentos, oferecendo uma visão complementar e resumida da mesma droga. Para a proteína, um potente modelo do tipo linguagem transforma cada aminoácido em um vetor sensível ao contexto, que é então analisado por vários filtros unidimensionais de tamanhos diferentes para captar motivos locais e padrões de maior alcance ao longo da sequência.
Ensinando o Modelo a Notar a Comunicação Cruzada
Ver drogas e proteínas separadamente não é suficiente; o essencial é como elas se influenciam quando se encontram. O MSCMF-DTB usa um mecanismo de cross-attention para que a representação da droga possa “olhar” seletivamente para diferentes regiões da proteína, e vice‑versa. Isso permite que o modelo destaque quais átomos e quais segmentos da proteína têm maior probabilidade de interagir. Além disso, uma rede tensorial especializada aprende combinações de ordem superior e não lineares de recursos de droga e proteína, indo além da simples mistura de características. As saídas do ramo de grafos, do ramo de fingerprint, do ramo protéico e do ramo de interação são então mescladas e passadas para um módulo final de predição que pode executar tanto a classificação binária de ligação (sim/não) quanto a pontuação contínua de afinidade.
Colocando o Sistema à Prova
Os autores avaliaram o MSCMF-DTB em um amplo conjunto de coleções benchmark amplamente usadas na área. Para decisões de ligação sim/não, testaram em cinco conjuntos cobrindo humanos, uma espécie de verme modelo, uma família de receptores de sinalização e grandes catálogos droga–proteína como BioSNAP e DrugBank. Para a força de ligação, usaram DAVIS e KIBA, dois conjuntos padrão focados em quinases que contêm valores medidos de afinidade. Em coleções pequenas e grandes, o modelo mostrou desempenho forte e consistente, frequentemente igualando ou superando métodos líderes. No grande conjunto DrugBank, por exemplo, ele melhorou a área sob a curva ROC em até 3,2% e o recall em 6,1% em comparação com o modelo anterior de melhor desempenho, significando que encontrou mais interações verdadeiras sem colapsar com o tamanho e a diversidade dos dados.
Espiando por Dentro e Testando um Cenário Realista
Para garantir que o modelo não seja apenas uma caixa preta, os pesquisadores examinaram onde seu mecanismo de atenção se concentrava em estruturas proteicas reais com sítios de ligação a drogas conhecidos. Quando mapearam as regiões de alta atenção em modelos tridimensionais de proteínas, muitos dos resíduos destacados sobrepuseram‑se às regiões de ligação confirmadas experimentalmente, sugerindo que o sistema está aprendendo sinais biologicamente relevantes. Eles também executaram um experimento exigente de “cold-start” no alvo relacionado ao câncer AKT1, excluindo completamente essa proteína do treinamento. Ao pedir para ranquear milhares de compostos candidatos, o modelo colocou vários inibidores conhecidos de AKT1 entre as principais previsões, mostrando que pode generalizar para alvos previamente não vistos e apoiar cenários de triagem virtual e reposicionamento.
O Que Isso Significa para Medicamentos Futuros
Em termos simples, o MSCMF-DTB é um casamenteiro de múltiplas visões entre drogas e proteínas. Ao combinar grafos atômicos detalhados, estatísticas de fragmentos e contexto rico de sequências protéicas, e ao modelar explicitamente como essas peças se comunicam, ele oferece previsões mais precisas e estáveis do que muitas abordagens existentes. Embora não substitua experimentos laboratoriais, pode reduzir significativamente a lista de candidatos promissores e sugerir onde contatos importantes podem se formar. Em conjunto com simulações baseadas em física e validação experimental, estruturas como o MSCMF-DTB podem ajudar a tornar a longa e cara jornada de molécula a medicamento mais rápida, barata e mais informada.
Citação: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Palavras-chave: interação droga–alvo, aprendizado profundo na descoberta de fármacos, triagem virtual, predição de afinidade de ligação, fusão cross-modal