Clear Sky Science · sv
MSCMF-DTB: en mångskalig tvärmodal fusionsram för prediktion av läkemedels–målbindning
Varför smartare matchning av läkemedel spelar roll
Att ta reda på vilka läkemedel som binder till vilka proteiner i våra kroppar liknar att lägga ett gigantiskt 3D-pussel med miljontals bitar. Att testa varje möjligt läkemedels–proteinpar i labbet är långsamt och dyrt, så forskare vänder sig till artificiell intelligens för att begränsa sökningen. Denna artikel presenterar MSCMF-DTB, ett nytt djuplärandesystem som är utformat för att förutsäga både om ett läkemedel kommer att binda till ett protein och hur starkt bindningen blir. Genom att kombinera flera typer av molekylär information i en och samma modell syftar det till att snabba upp läkemedelsupptäckt, vägleda läkemedelsomplacering och göra virtuell screening mer pålitlig.
Att betrakta läkemedel och proteiner från många vinklar
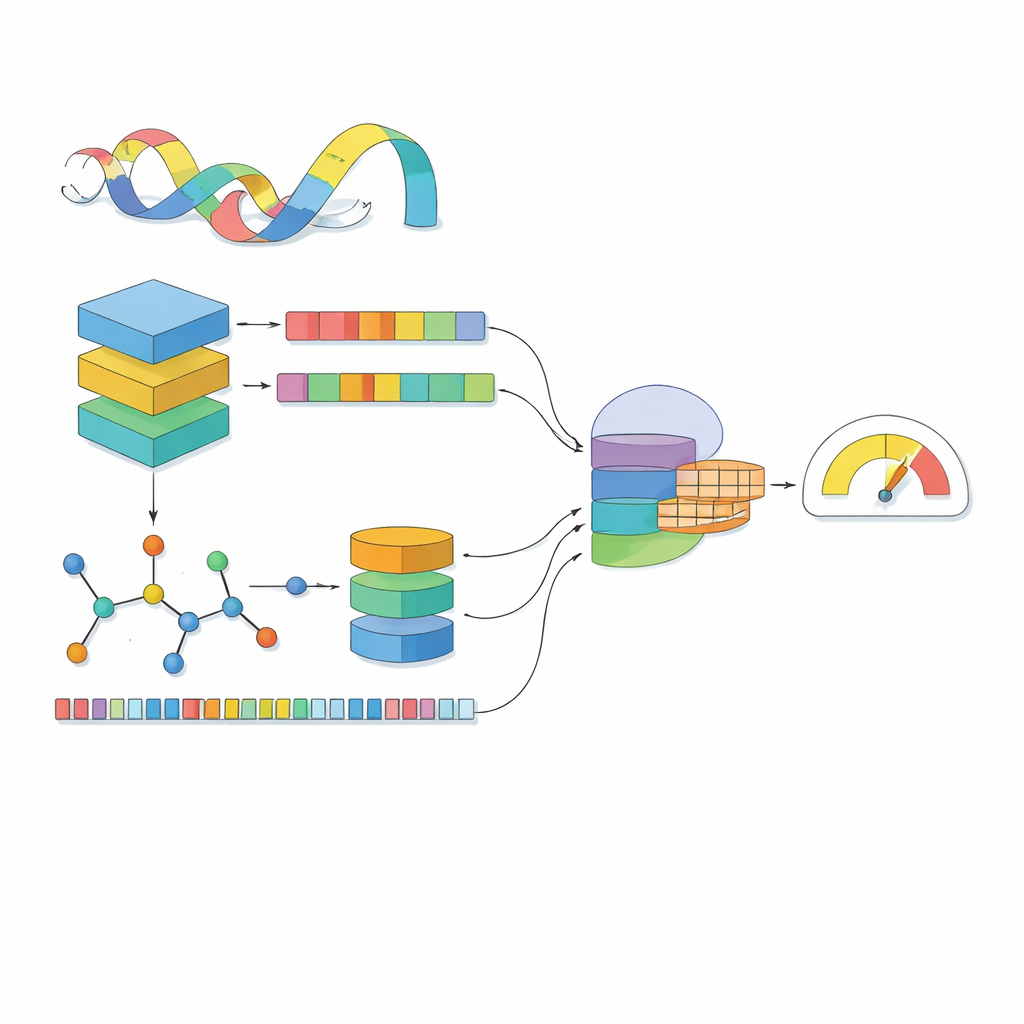
De flesta datormodeller ser ett läkemedel som en rad symboler och ett protein som en sträng av bokstäver. MSCMF-DTB går längre genom att betrakta varje läkemedel både som ett nätverk av atomer och bindningar och som ett mönster av återkommande kemiska fragment, samtidigt som proteinet behandlas som en sekvens berikad med kontext lärd från miljontals naturliga proteiner. För läkemedlet bygger modellen en graf där atomer är noder och bindningar länkar, och skickar denna graf genom ett djupt nätverk som gradvis lär sig mönster från små grannskap upp till hela molekylen. Parallellt fångar en andra kanal hur ofta vissa atomtyper och fragment förekommer, vilket erbjuder en kompletterande, sammanfattande vy av samma läkemedel. För proteinet omvandlar en kraftfull språkliknande modell först varje aminosyra till en kontextmedveten vektor, vilken sedan skannas av flera endimensionella filter i olika storlekar för att plocka upp lokala motiv och långräckviddsmönster längs sekvensen.
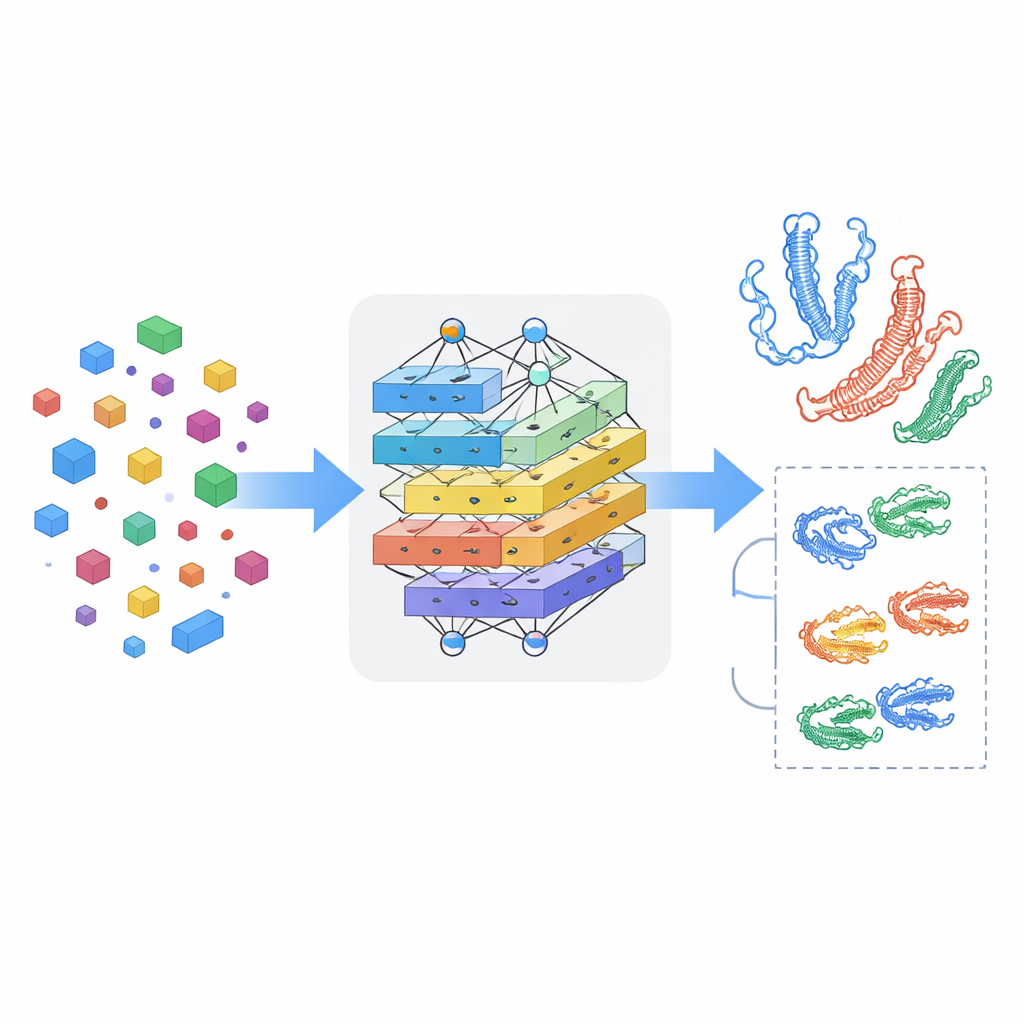
Lära modellen att lägga märke till ömsesidig påverkan
Att se läkemedel och proteiner var för sig räcker inte; kärnan är hur de påverkar varandra när de möts. MSCMF-DTB använder en cross-attention-mekanism så att läkemedlets representation selektivt kan ”titta på” olika regioner av proteinet, och vice versa. Detta gör det möjligt för modellen att lyfta fram vilka atomer och vilka proteinsegment som sannolikt interagerar. Utöver detta lär ett specialiserat tensornätverk högre ordningens, icke-linjära kombinationer av läkemedels- och proteinfunktioner, vilket går längre än enkel funktionsblandning. Utdata från grafgrenen, fingerprint-grenen, proteingrenen och interaktionsgrenen slås sedan samman och matas in i en slutlig prediktionsmodul som kan utföra antingen ja/nej-klassificering av bindning eller kontinuerlig affinitetsbedömning.
Sätta systemet på prov
Författarna utvärderade MSCMF-DTB på en bred uppsättning benchmark-samlingar som är vanligt använda inom fältet. För bindnings ja/nej-beslut testade de på fem dataset som täcker människor, en modellmaskarart, en familj av signaleringsreceptorer och stora läkemedels–proteinkataloger som BioSNAP och DrugBank. För bindningsstyrka använde de DAVIS och KIBA, två standarddataset fokuserade på kinaser som innehåller uppmätta affinitetsvärden. Över små och stora samlingar visade modellen stark och konsekvent prestanda, ofta matchande eller överträffande ledande metoder. På det stora DrugBank-datasetet förbättrade den till exempel area under ROC-kurvan med upp till 3,2 % och recall med 6,1 % jämfört med den tidigare bästa modellen, vilket innebär att den hittade fler sanna interaktioner utan att kollapsa under datans storlek och mångfald.
En titt inuti och ett realistiskt test
För att försäkra sig om att modellen inte bara är en svart låda undersökte forskarna var dess attention-mekanism fokuserade på verkliga proteinstrukturer med kända läkemedelsbindande platser. När de kartlade de höga attenueringsregionerna på tredimensionella proteinmodeller överlappade många av de markerade resterna med experimentellt bekräftade bindningsregioner, vilket tyder på att systemet lär sig biologiskt meningsfulla signaler. De körde också ett krävande ”cold-start”-experiment på det cancerrelaterade målet AKT1, där detta protein helt uteslöts från träningen. När modellen ombads rangordna tusentals kandidatföreningar placerade den flera kända AKT1‑hämmare bland toppförutsägelserna, vilket visar att den kan generalisera till tidigare osedda mål och stödja scenarier för virtuell screening och omplacering.
Vad detta betyder för framtidens läkemedel
Enkelt uttryckt är MSCMF-DTB en multimodal matchmaker mellan läkemedel och proteiner. Genom att kombinera detaljerade atomära grafer, fragmentstatistik och rik proteinsekvenskontext, och genom att explicit modellera hur dessa delar kommunicerar med varandra, erbjuder den mer exakta och stabila förutsägelser än många befintliga angreppssätt. Även om den inte ersätter laboratorieexperiment kan den kraftigt begränsa listan över lovande kandidater och föreslå var viktiga kontakter kan bildas. Tillsammans med fysikbaserade simuleringar och experimentell validering kan ramverk som MSCMF-DTB hjälpa till att göra den långa och kostsamma resan från molekyl till läkemedel snabbare, billigare och mer informerad.
Citering: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Nyckelord: läkemedels–målsinteraktion, djuplärande i läkemedelsupptäckt, virtuell screening, prediktion av bindningsaffinitet, tvärmodal fusion