Clear Sky Science · it
MSCMF-DTB: un framework di fusione cross-modale multi-scala per la previsione del legame farmaco‑bersaglio
Perché è importante abbinare i farmaci con maggiore intelligenza
Scoprire quali farmaci si legano a quali proteine nel nostro corpo è come risolvere un gigantesco puzzle tridimensionale con milioni di pezzi. Testare in laboratorio ogni possibile coppia farmaco‑proteina è lento e costoso, quindi i ricercatori si rivolgono all’intelligenza artificiale per restringere la ricerca. Questo articolo presenta MSCMF-DTB, un nuovo sistema di deep learning progettato per prevedere sia se un farmaco si legherà a una proteina sia quanto fortemente lo farà. Combinando più tipi di informazioni molecolari in un unico modello, mira ad accelerare la scoperta di farmaci, guidare il riposizionamento di farmaci e rendere lo screening virtuale più affidabile.
Osservare farmaci e proteine da più angolazioni
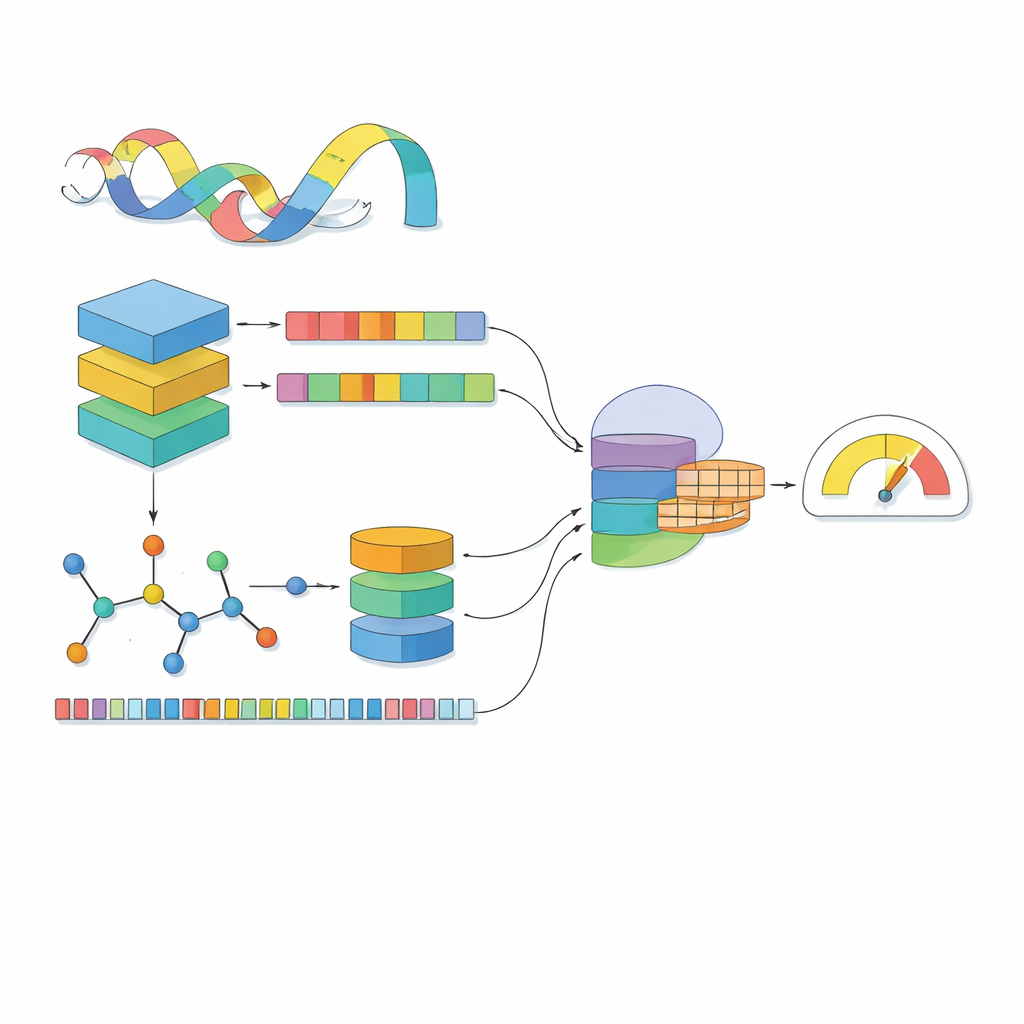
La maggior parte dei modelli computazionali vede un farmaco come una sequenza di simboli e una proteina come una stringa di lettere. MSCMF-DTB va oltre, considerando ogni farmaco sia come una rete di atomi e legami sia come un modello di frammenti chimici ricorrenti, mentre tratta la proteina come una sequenza arricchita da contesto appreso da milioni di proteine naturali. Per il farmaco, il modello costruisce un grafo in cui gli atomi sono nodi e i legami sono archi, poi fa passare questo grafo attraverso una rete profonda che impara progressivamente pattern dai vicini più piccoli fino all’intera molecola. In parallelo, un secondo canale cattura la frequenza di certi tipi di atomi e frammenti, offrendo una visione complementare e di sintesi dello stesso farmaco. Per la proteina, un potente modello simile a un linguaggio trasforma prima ogni amminoacido in un vettore sensibile al contesto, che viene poi esaminato da diversi filtri unidimensionali di varie dimensioni per cogliere motivi locali e schemi a lungo raggio lungo la sequenza.
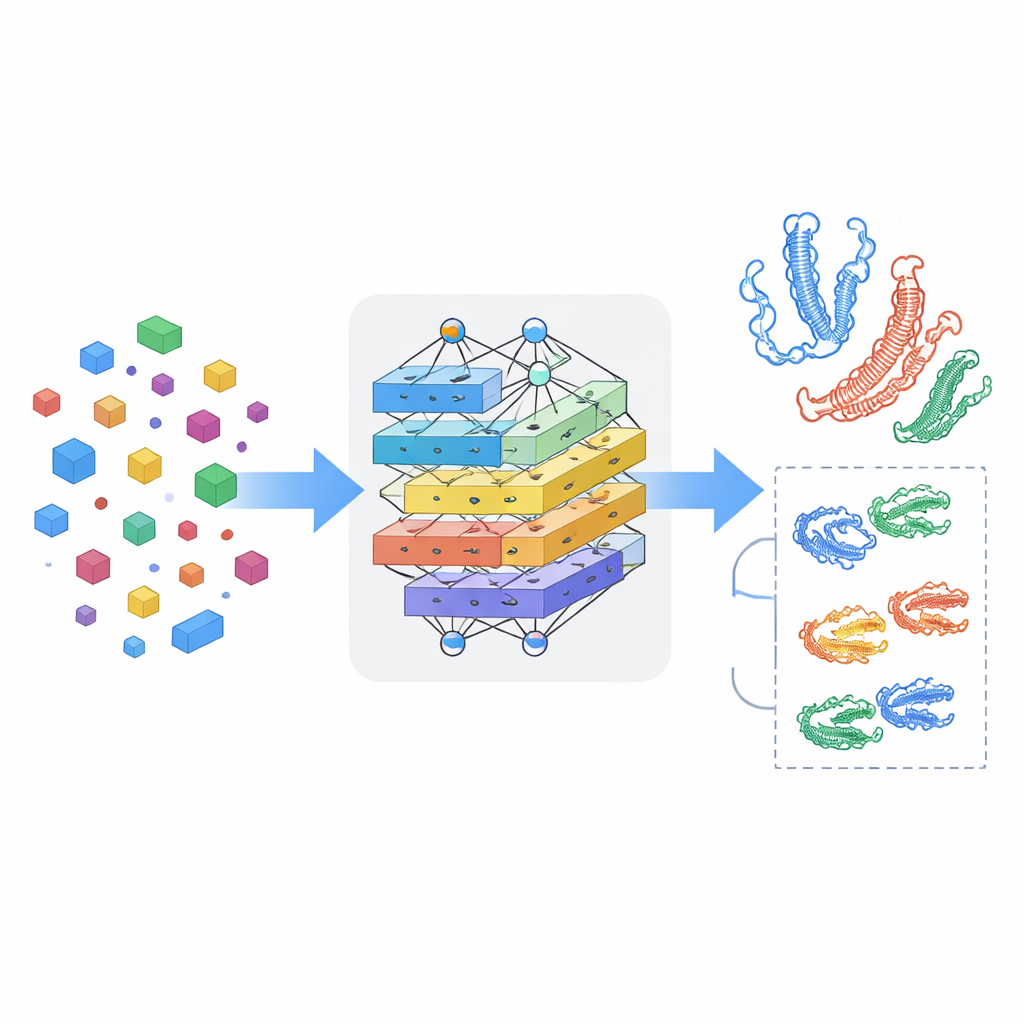
Insegnare al modello a cogliere il dialogo reciproco
Vedere farmaci e proteine separatamente non basta; la chiave è come si influenzano a vicenda quando si incontrano. MSCMF-DTB usa un meccanismo di cross-attention in modo che la rappresentazione del farmaco possa “guardare” selettivamente diverse regioni della proteina, e viceversa. Questo permette al modello di evidenziare quali atomi e quali segmenti proteici sono più probabilmente coinvolti nell’interazione. Oltretutto, una rete tensore specializzata apprende combinazioni di ordine superiore e non lineari di caratteristiche di farmaco e proteina, andando oltre la semplice mescolanza di feature. Le uscite del ramo grafo, del ramo fingerprint, del ramo proteina e del ramo interazione vengono poi fuse e passate a un modulo di predizione finale che può eseguire sia una classificazione binaria di legame sia una stima continua dell’affinità.
Mettere il sistema alla prova
Gli autori hanno valutato MSCMF-DTB su un ampio insieme di collezioni benchmark ampiamente usate nel campo. Per le decisioni binarie di legame, hanno testato su cinque dataset che coprono esseri umani, una specie modello di verme, una famiglia di recettori di segnalazione e grandi cataloghi farmaco‑proteina come BioSNAP e DrugBank. Per la forza del legame, hanno utilizzato DAVIS e KIBA, due dataset standard focalizzati sulle chinasi contenenti valori di affinità misurati. Su collezioni piccole e grandi, il modello ha mostrato prestazioni solide e coerenti, spesso allineandosi o superando i metodi di punta. Sul grande dataset DrugBank, per esempio, ha migliorato l’area sotto la curva ROC fino al 3,2% e il recall del 6,1% rispetto al precedente miglior modello, il che significa che ha identificato più interazioni vere senza collassare di fronte alla dimensione e alla diversità dei dati.
Guardare dentro e provare un test realistico
Per assicurarsi che il modello non sia solo una scatola nera, i ricercatori hanno esaminato su quali regioni delle strutture proteiche reali il suo meccanismo di attenzione si concentrava, in presenza di siti di legame noti. Quando hanno mappato le regioni ad alta attenzione su modelli tridimensionali delle proteine, molti residui evidenziati si sovrapponevano a regioni di legame confermate sperimentalmente, suggerendo che il sistema apprende segnali biologicamente significativi. Hanno inoltre eseguito un impegnativo esperimento di “cold‑start” sul bersaglio correlato al cancro AKT1, escludendo completamente questa proteina dall’addestramento. Quando gli è stato chiesto di classificare migliaia di composti candidati, il modello ha posizionato diversi inibitori noti di AKT1 tra le prime previsioni, dimostrando di sapersi generalizzare a bersagli precedentemente non visti e di poter supportare scenari di screening virtuale e riposizionamento.
Cosa significa questo per i farmaci del futuro
In termini semplici, MSCMF-DTB è un abbinatore multi‑vista tra farmaci e proteine. Combinando grafi atomici dettagliati, statistiche sui frammenti e un ricco contesto di sequenza proteica, e modellando esplicitamente come questi elementi dialogano tra loro, offre previsioni più accurate e stabili rispetto a molti approcci esistenti. Pur non sostituendo gli esperimenti di laboratorio, può ridurre notevolmente la lista di candidati promettenti e suggerire dove potrebbero formarsi i contatti importanti. Accoppiato con simulazioni basate sulla fisica e con la validazione sperimentale, framework come MSCMF-DTB potrebbero rendere il lungo e costoso percorso da molecola a medicina più rapido, meno caro e più informato.
Citazione: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Parole chiave: interazione farmaco‑bersaglio, deep learning nella scoperta di farmaci, screening virtuale, predizione dell’affinità di legame, fusione cross-modale