Clear Sky Science · es
MSCMF-DTB: un marco de fusión multimodal a varias escalas para la predicción de unión fármaco‑diana
Por qué importa emparejar fármacos con más inteligencia
Determinar qué medicamentos se unen a qué proteínas dentro de nuestro organismo es como resolver un gigantesco rompecabezas 3D con millones de piezas. Probar en el laboratorio cada posible par fármaco‑proteína es lento y caro, por lo que los investigadores recurren a la inteligencia artificial para acotar la búsqueda. Este artículo presenta MSCMF-DTB, un nuevo sistema de aprendizaje profundo diseñado para predecir tanto si un fármaco se unirá a una proteína como la intensidad de esa unión. Al combinar varios tipos de información molecular en un único modelo, pretende acelerar el descubrimiento de fármacos, orientar la reprofilación de fármacos y hacer que el cribado virtual sea más fiable.
Mirando fármacos y proteínas desde múltiples ángulos
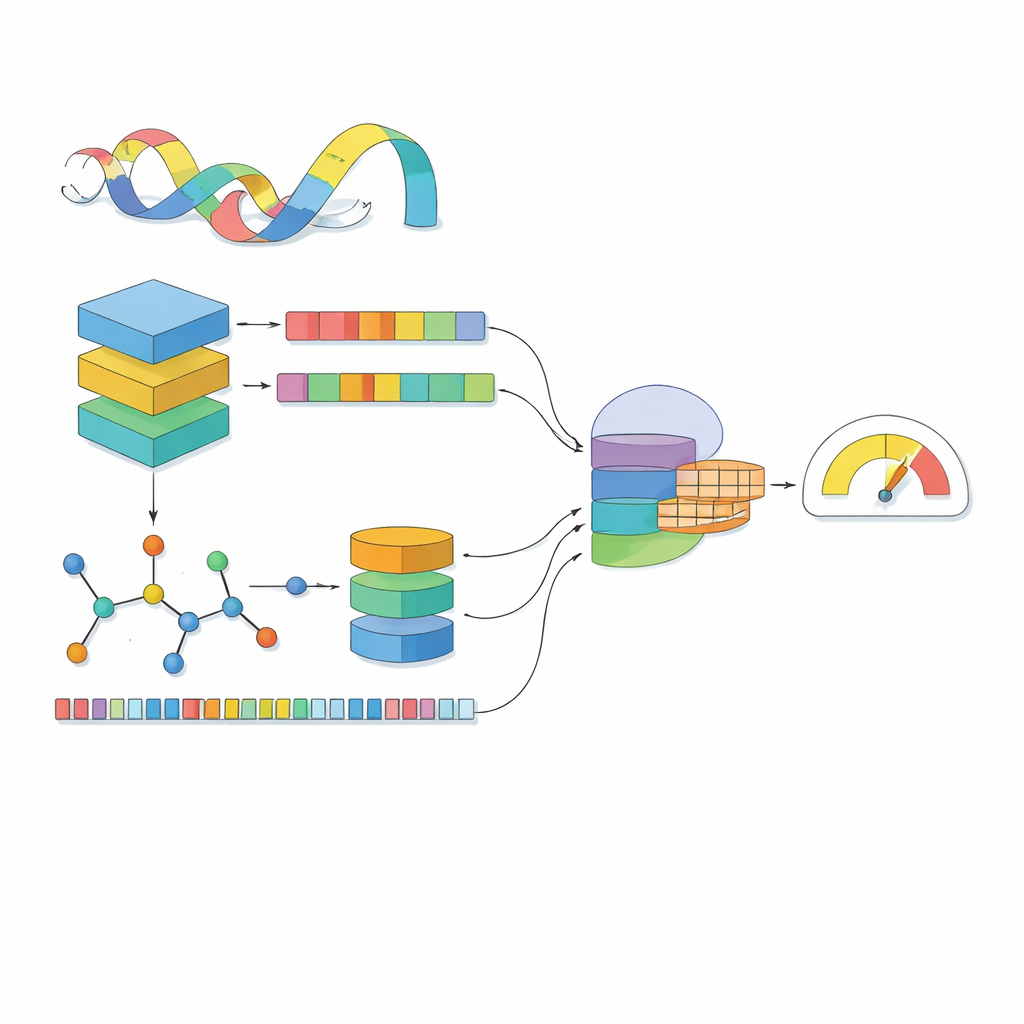
La mayoría de los modelos informáticos ven un fármaco como una línea de símbolos y una proteína como una cadena de letras. MSCMF-DTB va más allá al considerar cada fármaco tanto como una red de átomos y enlaces como un patrón de fragmentos químicos recurrentes, mientras trata la proteína como una secuencia enriquecida con contexto aprendido a partir de millones de proteínas naturales. Para el fármaco, el modelo construye un grafo donde los átomos son nodos y los enlaces son aristas, y luego procesa ese grafo mediante una red profunda que aprende progresivamente patrones desde vecindades pequeñas hasta la molécula completa. En paralelo, un segundo canal captura la frecuencia de aparición de ciertos tipos de átomos y fragmentos, ofreciendo una visión complementaria y resumida del mismo fármaco. Para la proteína, un potente modelo de tipo lingüístico convierte primero cada aminoácido en un vector con conocimiento contextual, que a continuación es escaneado por varios filtros unidimensionales de distinto tamaño para detectar motivos locales y patrones de mayor alcance a lo largo de la secuencia.
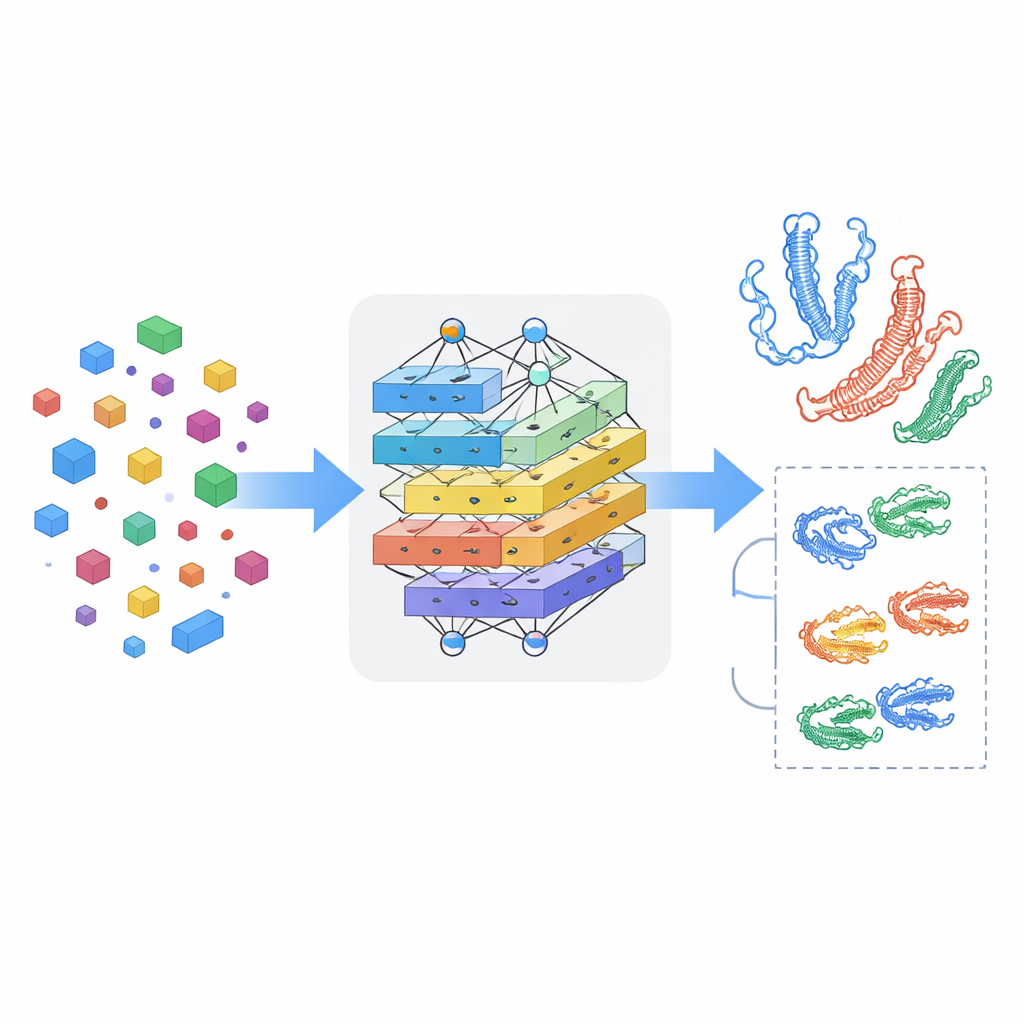
Enseñar al modelo a captar la interacción mutua
Ver fármacos y proteínas por separado no basta; lo esencial es cómo se influyen cuando se encuentran. MSCMF-DTB utiliza un mecanismo de atención cruzada para que la representación del fármaco pueda “mirar” selectivamente distintas regiones de la proteína, y viceversa. Esto permite al modelo resaltar qué átomos y qué segmentos proteicos tienen mayor probabilidad de interactuar. Además, una red tensorial especializada aprende combinaciones de orden superior y no lineales de las características de fármaco y proteína, yendo más allá de una simple mezcla de rasgos. Las salidas de la rama de grafos, la rama de huellas (fingerprint), la rama proteica y la rama de interacción se fusionan y pasan a un módulo de predicción final capaz de realizar tanto clasificación binaria de unión sí/no como estimación continua de afinidad.
Poner el sistema a prueba
Los autores evaluaron MSCMF-DTB en un conjunto amplio de colecciones de referencia muy utilizadas en el campo. Para las decisiones de unión sí/no, realizaron pruebas en cinco conjuntos de datos que abarcan humanos, una especie modelo de gusano, una familia de receptores de señalización y grandes catálogos fármaco‑proteína como BioSNAP y DrugBank. Para la intensidad de unión, emplearon DAVIS y KIBA, dos conjuntos de datos estándar centrados en quinasas con valores de afinidad medidos. En colecciones pequeñas y grandes, el modelo mostró un desempeño fuerte y consistente, igualando o superando con frecuencia a los métodos líderes. En el gran conjunto DrugBank, por ejemplo, mejoró el área bajo la curva ROC hasta en un 3,2 % y el recall en un 6,1 % respecto al modelo previo mejor, lo que significa que encontró más interacciones verdaderas sin colapsar ante el tamaño y la diversidad de los datos.
Mirar dentro del modelo y probar un escenario realista
Para asegurarse de que el modelo no es solo una caja negra, los investigadores examinaron dónde se centraba su mecanismo de atención en estructuras proteicas reales con sitios de unión conocidos. Al mapear las regiones de alta atención sobre modelos proteicos tridimensionales, muchos de los residuos resaltados se solaparon con regiones de unión confirmadas experimentalmente, lo que sugiere que el sistema aprende señales biológicamente significativas. También realizaron un exigente experimento de “arranque en frío” en el objetivo relacionado con el cáncer AKT1, excluyendo completamente esa proteína del entrenamiento. Al pedir al modelo que ordenara miles de compuestos candidatos, situó varios inhibidores conocidos de AKT1 entre las principales predicciones, demostrando que puede generalizar a dianas no vistas previamente y apoyar escenarios de cribado virtual y reprofilación.
Qué supone esto para los futuros medicamentos
En términos sencillos, MSCMF-DTB es un casamentero multimodal entre fármacos y proteínas. Al combinar grafos atómicos detallados, estadísticas de fragmentos y un rico contexto de la secuencia proteica, y modelar explícitamente cómo estas piezas se comunican entre sí, ofrece predicciones más precisas y estables que muchas aproximaciones existentes. Aunque no sustituye a los experimentos de laboratorio, puede reducir considerablemente la lista de candidatos prometedores y sugerir dónde pueden formarse contactos importantes. Unido a simulaciones basadas en la física y a la validación experimental, marcos como MSCMF-DTB podrían ayudar a que el largo y costoso camino de la molécula al medicamento sea más rápido, barato y mejor informado.
Cita: Huang, J., Pan, Y. & Chen, Q. MSCMF-DTB: a multi-scale cross-modal fusion framework for drug–target binding prediction. Sci Rep 16, 13211 (2026). https://doi.org/10.1038/s41598-026-44048-9
Palabras clave: interacción fármaco‑diana, aprendizaje profundo en descubrimiento de fármacos, cribado virtual, predicción de afinidad de unión, fusión cruzada modal