Clear Sky Science · sv
Utveckling av en pKa-prediktor (pKaLearn) genom att utnyttja undervisningserfarenhet för att förbättra maskininlärning
Varför det är viktigt att lära maskiner om syror
Vardagsprodukter från läkemedel till batterier är beroende av hur lätt molekyler avger eller tar upp protoner, en egenskap som fångas av den bekanta pH-skalan och dess molekylära kusin pKa. Att mäta pKa i labbet tar tid och arbete, och även avancerade datorbaserade metoder kan vara långsamma eller opålitliga. Denna studie ställer en enkel fråga med stora följder: tänk om vi lärde datorer grundläggande kemi på samma sätt som vi lär studenter, och sedan bad dem förutsäga pKa-värden snabbare och mer exakt?
Från klassrumsregler till datorregler
I kemilektioner lär sig studenter steg för steg. Först kommer atomer och enkla trender som elektronegativitet, sedan idéer som resonans, ringspänning och hur närliggande grupper drar eller skjuter elektroner. Först senare tar man itu med knepiga förutsägelser, som vilken väteatom i en komplex molekyl som blir mest sur. Författarna menar att de flesta maskininlärningsmodeller hoppar över denna progression. De matas ofta med råa strukturer eller abstrakta fingeravtryck och får upptäcka mönster på egen hand, vilket kan få dem att memorerar exempel istället för att förstå bakomliggande orsaker. Här kodar teamet medvetet in samma grundläggande idéer som mänskliga lärare använder och matar in dem i sina algoritmer.
Bygga en modell som tänker som en kemist
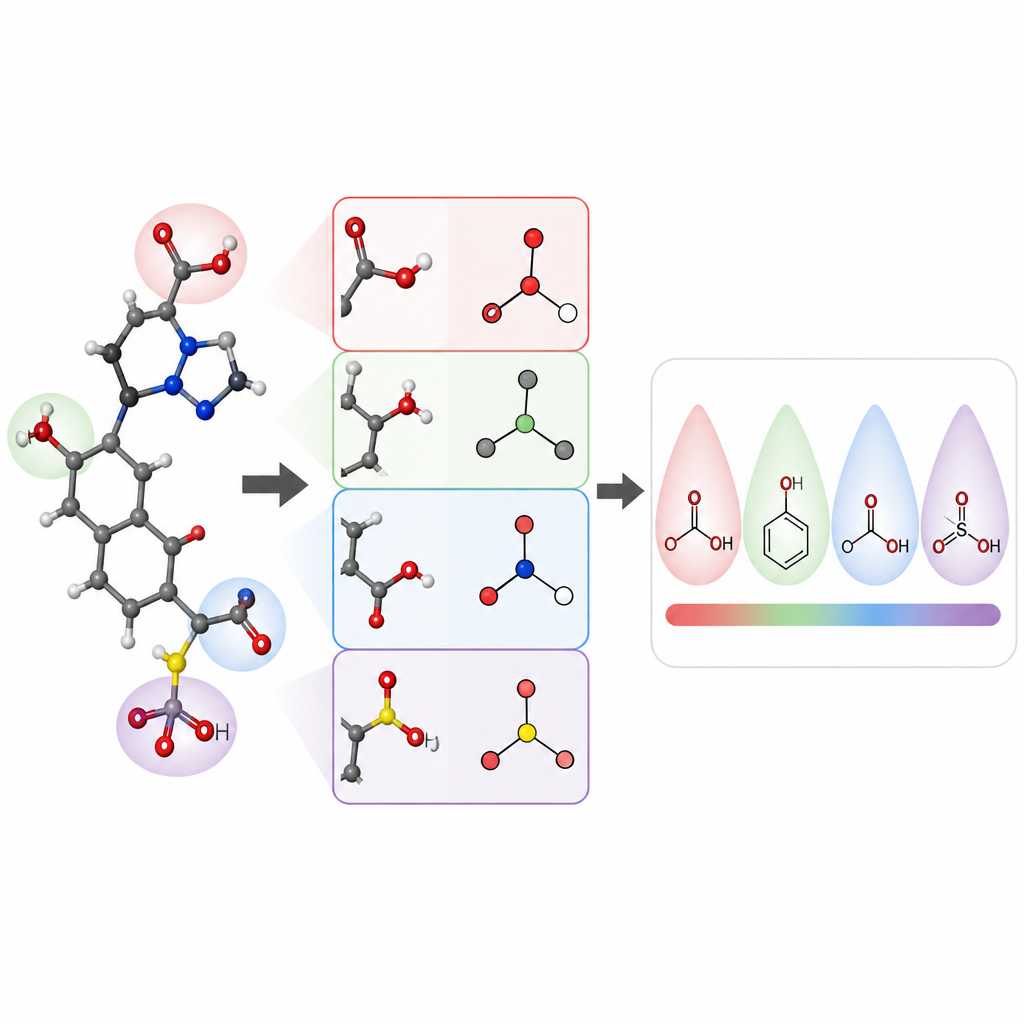
Forskarlaget fokuserade på att förutsäga pKa-värden för små organiska molekyler, en kärnegenskap för läkemedelsdesign och många områden inom kemin. De byggde en modell kallad pKaLearn med ett grafneuronätverk, som behandlar en molekyl som en uppsättning atomer kopplade av bindningar. Istället för att förlita sig enbart på generiska strukturella koder lade de till egenskaper knutna direkt till läroboksprinciper: hur polar en bindning är, hur många bindningar som skiljer en laddad grupp från en joniserbar plats, om en bindning är en del av en ring och hur konjugation och resonans kan sprida laddning. De mätte också hur långt subtila effekter, som induktivt drag, färdas längs en atomkedja och utformade nätverket så att varje joniserbart ställe ”känner” sin omgivning upp till ungefär sju bindningar bort.
Undervisning, testning och att undvika enkel memorering
För att kontrollera om deras undervisningsstil verkligen hjälpte datorn att lära sig, satte författarna noggrant samman och rensade en datamängd på ungefär 13 000 pKa-värden. Istället för den vanliga slumpmässiga uppdelningen, som kan göra tränings- och testmolekyler mycket lika, klustrade de molekyler så att testuppsättningen skulle innehålla nya kemiska familjer. Detta svårare prov avslöjar om modellen lärt sig generella regler eller helt enkelt memorerat exempel. Under dessa förhållanden uppnådde pKaLearn typiska fel under 0,7 pKa-enheter, bättre än traditionella statistiska modeller baserade på fingeravtryck och starkare än flera befintliga maskininlärnings- och kvantkemi-baserade prediktorer. När de provade vanliga genvägar, såsom att förlita sig endast på elementtyper eller en vanlig programvarudefinition av konjugerade bindningar, sjönk prestandan, vilket betonar värdet av kemiskt meningsfulla, väldefinierade egenskaper.
Jämfört med andra smarta prediktorer
Teamet jämförde pKaLearn med välkända pKa-verktyg på mycket använda benchmarkuppsättningar, inklusive molekyler från läkemedelsföretag och blinda förutsägelseutmaningar där de verkliga svaren var dolda under utvecklingen. I dessa tester matchade eller överträffade deras modell konsekvent metoder som kombinerar tunga kvantberäkningar med maskininlärning, liksom andra grafbaserade neurala nätverk. Viktigt är att de också undersökte fall där förutsägelser gick fel, ofta ochspårande dem till saknade joniserbara platser, knepiga tautomerer som kan flytta protoner inom en molekyl, eller experimentdata som i sig var tvetydiga. Sammantaget visade endast en liten andel föreningar stora fel, och prestandan förblev stabil över många olika funktionella grupper.
Vad detta betyder för kemister och längre än så
Studien visar att maskiner kan dra nytta av att bli undervisade som studenter. Genom att bädda in enkla men kraftfulla kemiska idéer direkt i en inlärningsalgoritm skapade författarna en pKa-prediktor som både är noggrann och mer tolkbar än svartlådemodeller. För praktiska användare innebär detta snabbare, mer pålitliga uppskattningar av surhets- och basstyrka över ett brett spektrum av molekyler, vilket hjälper till att vägleda läkemedelsdesign och andra molekylupptäcktsinsatser. Mer allmänt pekar arbetet på en väg för framtida verktyg: istället för att be artificiell intelligens återskapa grundläggande vetenskap från grunden, kan vi bygga in mänsklig expertis och låta modellerna fokusera på att förfina och utvidga den kunskapen.
Citering: Genzling, J., Luo, Z., Weiser, B. et al. Development of a pKa predictor (pKaLearn) by leveraging teaching experience to improve machine learning. Commun Chem 9, 181 (2026). https://doi.org/10.1038/s42004-026-01983-y
Nyckelord: pKa-förutsägelse, maskininlärning, grafneuronätverk, beräkningskemi, Läkemedelsdesign