Clear Sky Science · en
Development of a pKa predictor (pKaLearn) by leveraging teaching experience to improve machine learning
Why teaching machines about acids matters
Everyday products from medicines to batteries rely on how easily molecules give up or accept protons, a property captured by the familiar pH scale and its molecular cousin, pKa. Measuring pKa in the lab takes time and effort, and even advanced computer methods can be slow or unreliable. This study asks a simple question with big implications: what if we taught computers basic chemistry the way we teach students, then asked them to predict pKa values faster and more accurately?
From classroom rules to computer rules
In chemistry class, students learn step by step. First come atoms and simple trends like electronegativity, then ideas such as resonance, ring strain, and how nearby groups pull or push electrons. Only later do they tackle tricky predictions, like which hydrogen in a complex molecule will be most acidic. The authors argue that most machine learning models skip this progression. They are often fed raw structures or abstract fingerprints and left to discover patterns alone, which can cause them to memorize examples instead of understanding the underlying causes. Here, the team deliberately encodes the same basic ideas human teachers use and feeds those into their algorithms.
Building a model that thinks like a chemist
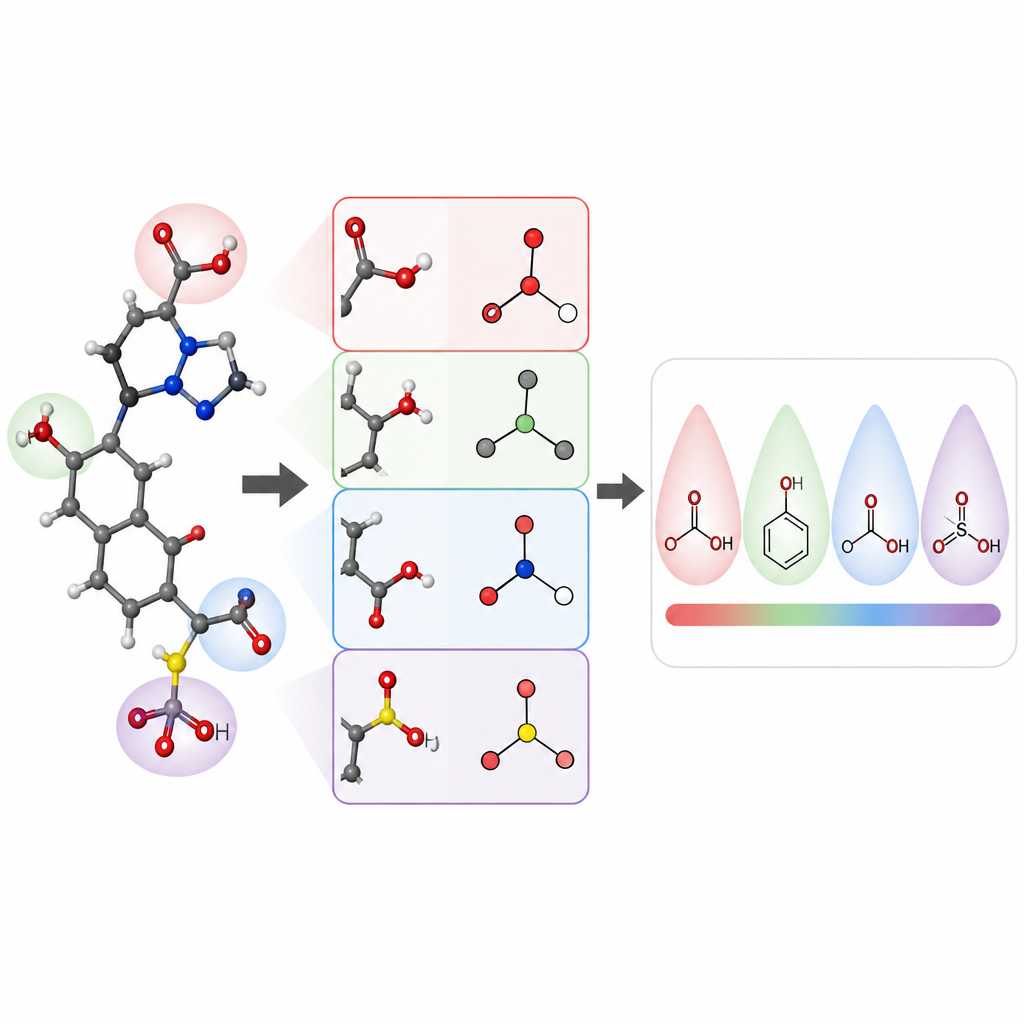
The researchers focused on predicting pKa values of small organic molecules, a core property for drug design and many areas of chemistry. They built a model called pKaLearn using a graph neural network, which treats a molecule as a set of atoms connected by bonds. Instead of relying only on generic structural codes, they added features tied directly to textbook principles: how polar a bond is, how many bonds separate a charged group from an ionizable site, whether a bond is part of a ring, and how conjugation and resonance can spread charge. They also measured how far subtle effects such as inductive pull travel along a chain of atoms and designed the network so that each ionizable site “feels” its environment out to about seven bonds away.
Teaching, testing, and avoiding simple memorization
To check whether their teaching style really helped the computer learn, the authors carefully assembled and cleaned a dataset of about 13,000 pKa values. Instead of the usual random split, which can make training and test molecules very similar, they clustered molecules so that the test set would contain new chemical families. This harder exam reveals whether the model has learned general rules or simply memorized examples. Under these conditions, pKaLearn achieved typical errors below 0.7 pKa units, better than traditional statistical models based on fingerprints and stronger than several existing machine learning and quantum chemistry based predictors. When they tried standard shortcuts, such as relying only on element types or a common software definition of conjugated bonds, performance dropped, underscoring the value of chemically meaningful, well defined features.
Stacking up against other smart predictors
The team compared pKaLearn with well known pKa tools on widely used benchmark sets, including molecules from pharmaceutical companies and blind prediction challenges where the true answers were hidden during development. In these tests, their model consistently matched or outperformed methods that combine heavy quantum calculations with machine learning, as well as other graph based neural networks. Importantly, they also examined cases where predictions went wrong, often tracing them to missing ionizable sites, tricky tautomers that can shuffle protons inside a molecule, or experimental data that were themselves ambiguous. Overall, only a small fraction of compounds showed large errors, and performance remained steady across many different functional groups.
What this means for chemists and beyond
The study shows that machines can benefit from being taught like students. By embedding simple but powerful chemistry ideas directly into a learning algorithm, the authors created a pKa predictor that is both accurate and more interpretable than black box models. For practical users, this means faster, more reliable estimates of acidity and basicity across a broad range of molecules, helping guide drug design and other molecular discovery efforts. More broadly, the work suggests a path for future tools: instead of asking artificial intelligence to rediscover fundamental science from scratch, we can build in human expertise and let models focus on refining and extending that knowledge.
Citation: Genzling, J., Luo, Z., Weiser, B. et al. Development of a pKa predictor (pKaLearn) by leveraging teaching experience to improve machine learning. Commun Chem 9, 181 (2026). https://doi.org/10.1038/s42004-026-01983-y
Keywords: pKa prediction, machine learning, graph neural network, computational chemistry, drug design