Clear Sky Science · de
Entwicklung eines pKa‑Prädiktors (pKaLearn) durch Nutzung von Lehrerfahrung zur Verbesserung des maschinellen Lernens
Warum es wichtig ist, Maschinen etwas über Säuren beizubringen
Alltägliche Produkte von Arzneimitteln bis zu Batterien hängen davon ab, wie leicht Moleküle Protonen abgeben oder aufnehmen — eine Eigenschaft, die durch die vertraute pH‑Skala und ihr molekulares Pendant pKa beschrieben wird. Die Messung von pKa im Labor erfordert Zeit und Aufwand, und selbst fortgeschrittene Computerverfahren können langsam oder unzuverlässig sein. Diese Studie stellt eine einfache Frage mit großen Auswirkungen: Was, wenn wir Computern die Grundlagen der Chemie so beibringen, wie wir sie Studierenden vermitteln, und sie dann bitten, pKa‑Werte schneller und genauer vorherzusagen?
Von Klassenraumregeln zu Computerregeln
Im Chemieunterricht lernen Studierende Schritt für Schritt. Zuerst kommen Atome und einfache Trends wie Elektronegativität, dann Konzepte wie Resonanz, Ringspannung und wie benachbarte Gruppen Elektronen ziehen oder drücken. Erst später beschäftigen sie sich mit schwierigen Vorhersagen, etwa welche Wasserstoff‑Atome in einem komplexen Molekül am stärksten sauer sind. Die Autoren argumentieren, dass die meisten Machine‑Learning‑Modelle diesen Lernverlauf überspringen. Häufig werden ihnen Rohstrukturen oder abstrakte Fingerprints übergeben, und sie müssen Muster allein entdecken, was dazu führen kann, dass sie Beispiele auswendig lernen, anstatt die zugrunde liegenden Ursachen zu verstehen. Hier kodiert das Team bewusst dieselben grundlegenden Ideen, die menschliche Lehrende verwenden, und speist diese Merkmale in ihre Algorithmen ein.
Ein Modell bauen, das wie ein Chemiker denkt
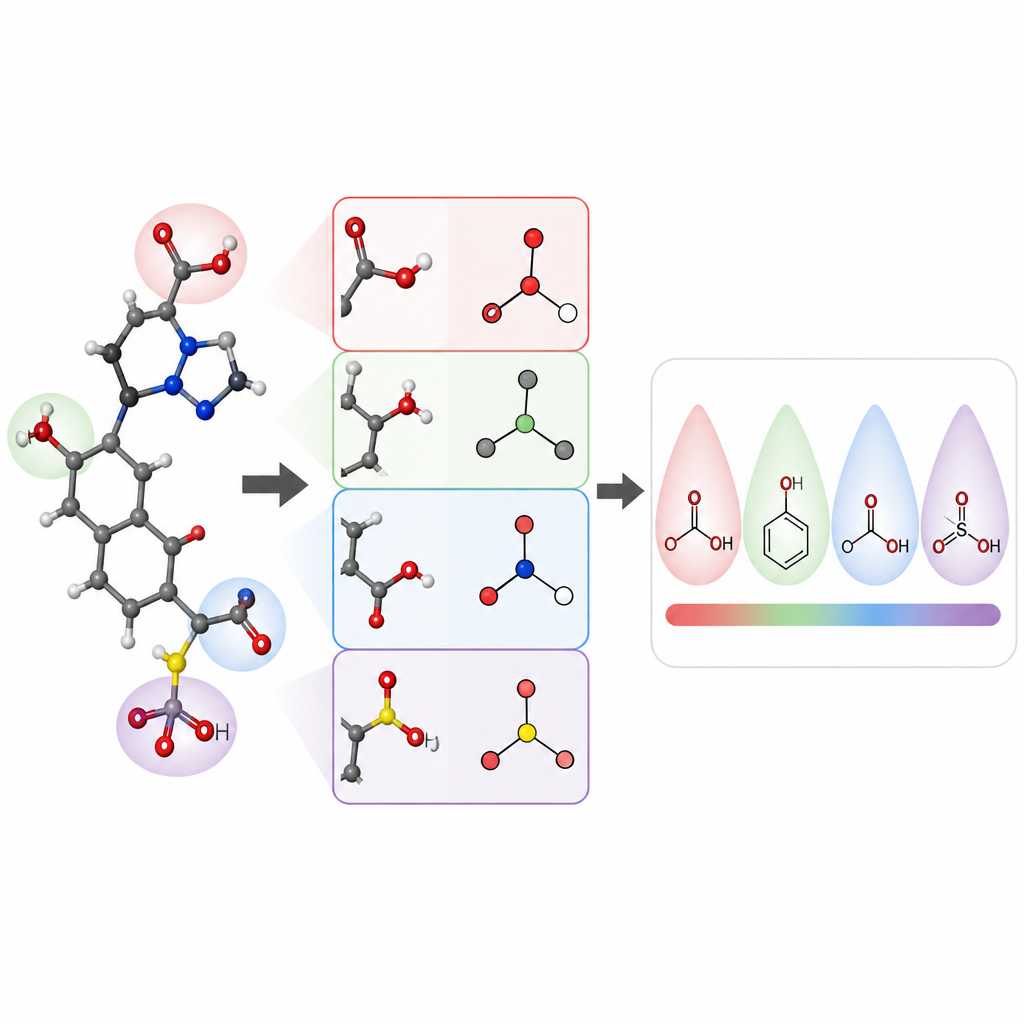
Die Forschenden konzentrierten sich auf die Vorhersage von pKa‑Werten kleiner organischer Moleküle, eine Kerngröße für das Wirkstoffdesign und viele Bereiche der Chemie. Sie entwickelten ein Modell namens pKaLearn auf Basis eines graph‑neuronal Netzwerks, das ein Molekül als Menge von Atomen betrachtet, die durch Bindungen verbunden sind. Anstatt sich nur auf generische Strukturcodes zu stützen, fügten sie Merkmale hinzu, die direkt an Lehrbuchprinzipien anknüpfen: wie polar eine Bindung ist, wie viele Bindungen eine geladene Gruppe von einer ionisierbaren Stelle trennen, ob eine Bindung Teil eines Rings ist und wie Konjugation und Resonanz Ladung verteilen können. Sie quantifizierten außerdem, wie weit subtile Effekte wie induktiver Zug entlang einer Atomenkette wirken, und entwarfen das Netzwerk so, dass jede ionisierbare Stelle ihre Umgebung bis zu etwa sieben Bindungen weit „spürt“.
Unterrichten, testen und einfaches Auswendiglernen vermeiden
Um zu prüfen, ob ihr Unterrichtsstil dem Computer wirklich beim Lernen hilft, stellten die Autoren sorgfältig einen Datensatz von etwa 13.000 pKa‑Werten zusammen und bereinigten ihn. Statt der üblichen zufälligen Aufteilung, die Trainings‑ und Testmoleküle sehr ähnlich machen kann, gruppierten sie Moleküle so, dass der Testdatensatz neue chemische Familien enthielt. Diese härtere Prüfung zeigt, ob das Modell allgemeine Regeln gelernt hat oder nur Beispiele auswendig gelernt wurden. Unter diesen Bedingungen erreichte pKaLearn typische Fehler unter 0,7 pKa‑Einheiten, besser als traditionelle statistische Modelle, die auf Fingerprints beruhen, und stärker als mehrere bestehende Machine‑Learning‑ und quantenchemische Prädiktoren. Als sie standardmäßige Abkürzungen ausprobierten, etwa die Beschränkung auf Elementtypen oder eine gebräuchliche Softwaredefinition konjugierter Bindungen, fiel die Leistung ab — ein Hinweis auf den Wert chemisch sinnvoller, gut definierter Merkmale.
Im Vergleich zu anderen intelligenten Prädiktoren
Das Team verglich pKaLearn mit bekannten pKa‑Tools auf weithin genutzten Benchmark‑Sätzen, darunter Moleküle aus Pharmaunternehmen und Blindtests, bei denen die wahren Antworten während der Entwicklung verborgen blieben. In diesen Prüfungen erreichte ihr Modell durchweg Leistungen auf Augenhöhe mit oder besser als Methoden, die auf aufwändigen quantenchemischen Berechnungen kombiniert mit maschinellem Lernen beruhen, sowie andere graphbasierte neuronale Netze. Wichtig ist, dass sie auch Fälle untersuchten, in denen Vorhersagen falsch lagen, und oft konnten sie die Fehler auf fehlende ionisierbare Stellen, knifflige Tautomere, die Protonen innerhalb eines Moleküls verschieben, oder auf experimentelle Daten zurückführen, die selbst mehrdeutig waren. Insgesamt zeigte nur ein kleiner Anteil der Verbindungen große Fehler, und die Leistung blieb über viele verschiedene funktionelle Gruppen hinweg stabil.
Was das für Chemiker und darüber hinaus bedeutet
Die Studie zeigt, dass Maschinen davon profitieren können, wie Studierende unterrichtet zu werden. Indem einfache, aber starke chemische Ideen direkt in einen Lernalgorithmus eingebettet werden, schufen die Autoren einen pKa‑Prädiktor, der sowohl genau als auch besser interpretierbar ist als Black‑Box‑Modelle. Für praktische Anwender bedeutet das schnellere, verlässlichere Schätzungen von Säure‑ und Basizitätswerten über ein breites Spektrum von Molekülen hinweg, was das Wirkstoffdesign und andere molekulare Entdeckungsprozesse unterstützt. Allgemeiner legt die Arbeit einen Weg für zukünftige Werkzeuge nahe: Anstatt die künstliche Intelligenz zu zwingen, grundlegende Wissenschaft von Grund auf neu zu entdecken, können wir menschliche Expertise einbauen und die Modelle darauf konzentrieren, dieses Wissen zu verfeinern und zu erweitern.
Zitation: Genzling, J., Luo, Z., Weiser, B. et al. Development of a pKa predictor (pKaLearn) by leveraging teaching experience to improve machine learning. Commun Chem 9, 181 (2026). https://doi.org/10.1038/s42004-026-01983-y
Schlüsselwörter: pKa‑Vorhersage, maschinelles Lernen, Graph‑Neurales Netzwerk, rechnergestützte Chemie, Wirkstoffdesign