Clear Sky Science · es
Desarrollo de un predictor de pKa (pKaLearn) aprovechando la experiencia docente para mejorar el aprendizaje automático
Por qué importa enseñar a las máquinas sobre ácidos
Productos cotidianos, desde medicamentos hasta baterías, dependen de lo fácil que es para las moléculas ceder o aceptar protones, una propiedad capturada por la familiar escala de pH y su análogo molecular, el pKa. Medir el pKa en el laboratorio exige tiempo y esfuerzo, e incluso métodos computacionales avanzados pueden ser lentos o poco fiables. Este estudio plantea una pregunta simple con grandes implicaciones: ¿y si enseñáramos a los ordenadores química básica de la misma forma que enseñamos a los estudiantes, y luego les pidiéramos que predigan valores de pKa más rápido y con mayor precisión?
De las reglas del aula a las reglas del ordenador
En las clases de química, los alumnos aprenden paso a paso. Primero vienen los átomos y tendencias simples como la electronegatividad, luego ideas como la resonancia, la tensión de anillo y cómo los grupos vecinos atraen o empujan electrones. Solo más adelante abordan predicciones complejas, como qué hidrógeno en una molécula complicada será el más ácido. Los autores sostienen que la mayoría de los modelos de aprendizaje automático se saltan esta progresión. Con frecuencia se les alimenta con estructuras crudas o huellas abstractas y se les deja descubrir patrones por sí solos, lo que puede llevarlos a memorizar ejemplos en lugar de comprender las causas subyacentes. Aquí, el equipo codifica deliberadamente las mismas ideas básicas que usan los docentes y las introduce en sus algoritmos.
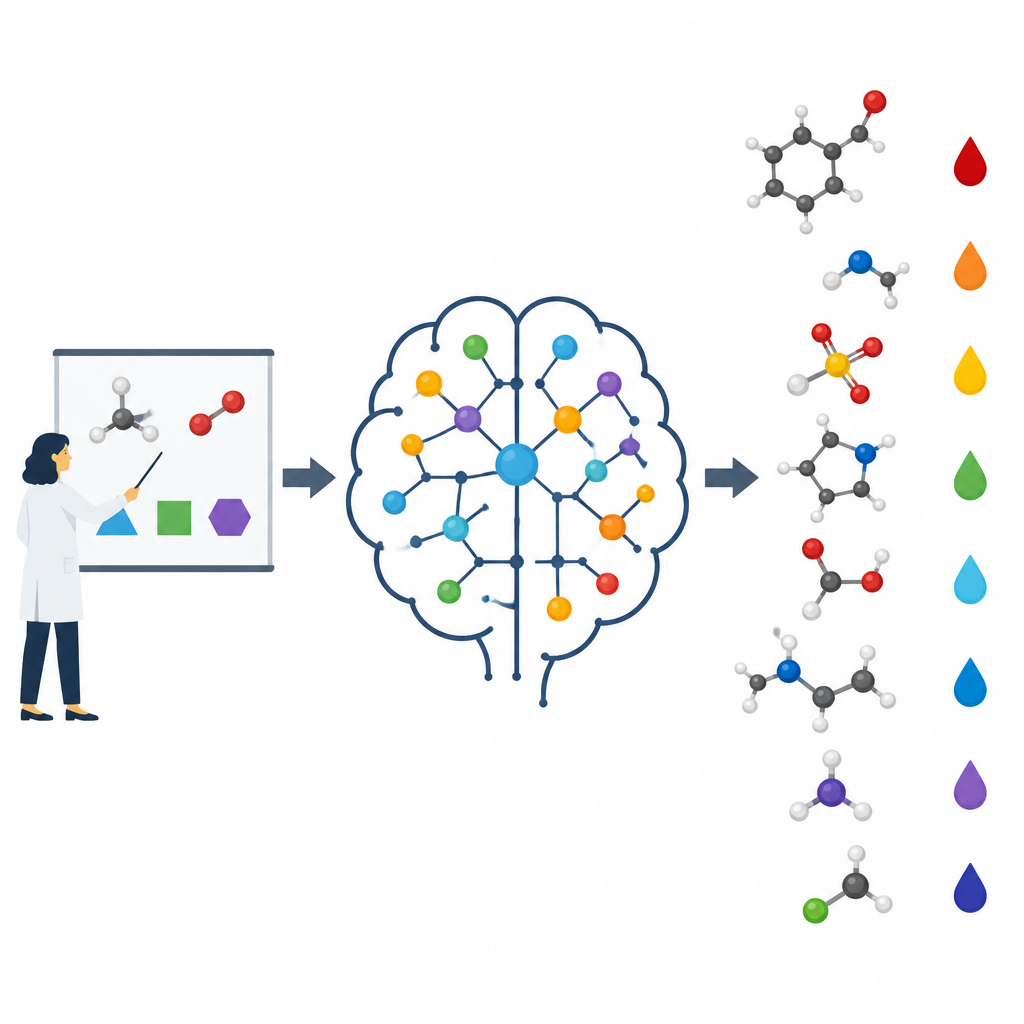
Construir un modelo que piense como un químico
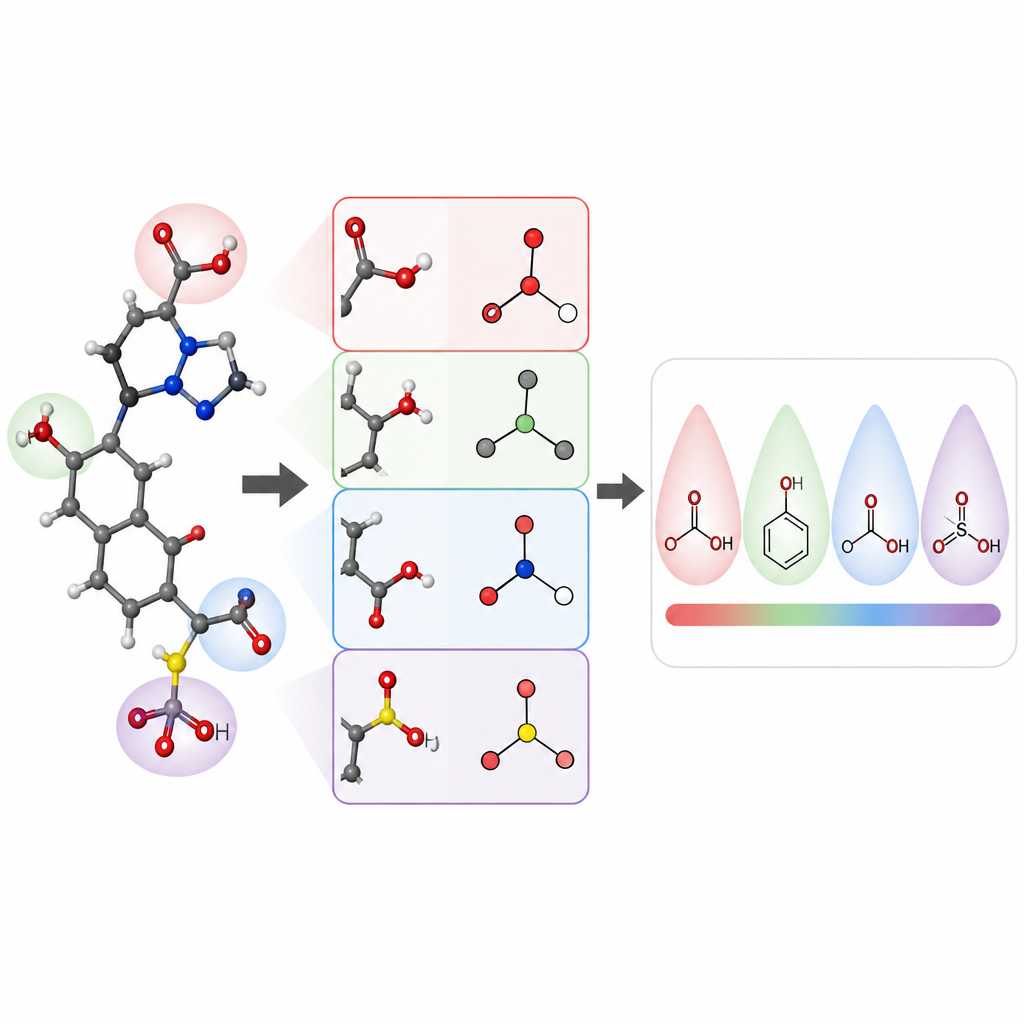
Los investigadores se centraron en predecir los valores de pKa de pequeñas moléculas orgánicas, una propiedad clave para el diseño de fármacos y muchas áreas de la química. Construyeron un modelo llamado pKaLearn usando una red neuronal gráfica, que trata a la molécula como un conjunto de átomos conectados por enlaces. En lugar de confiar solo en códigos estructurales genéricos, añadieron características vinculadas directamente a principios de libro de texto: cuán polar es un enlace, cuántos enlaces separan a un grupo cargado de un sitio ionizable, si un enlace forma parte de un anillo y cómo la conjugación y la resonancia pueden distribuir la carga. También midieron hasta qué distancia viajan efectos sutiles, como el arrastre inductivo a lo largo de una cadena de átomos, y diseñaron la red para que cada sitio ionizable “sienta” su entorno hasta aproximadamente siete enlaces de distancia.
Enseñar, evaluar y evitar la memorización simple
Para comprobar si su estilo de enseñanza realmente ayudaba al ordenador a aprender, los autores reunieron y limpiaron cuidadosamente un conjunto de datos de aproximadamente 13.000 valores de pKa. En lugar de la habitual división aleatoria, que puede hacer que las moléculas de entrenamiento y prueba sean muy similares, agruparon las moléculas para que el conjunto de prueba contuviera nuevas familias químicas. Este examen más exigente revela si el modelo ha aprendido reglas generales o simplemente ha memorizado ejemplos. Bajo estas condiciones, pKaLearn alcanzó errores típicos por debajo de 0,7 unidades de pKa, mejor que los modelos estadísticos tradicionales basados en huellas y superior a varios predictores existentes basados en aprendizaje automático y química cuántica. Cuando probaron atajos estándar, como confiar solo en tipos de elementos o en una definición común de software de enlaces conjugados, el rendimiento cayó, lo que subraya el valor de características químicamente significativas y bien definidas.
Compararlo con otros predictores avanzados
El equipo comparó pKaLearn con herramientas de pKa bien conocidas en conjuntos de referencia ampliamente usados, incluyendo moléculas de compañías farmacéuticas y desafíos de predicción a ciegas donde las respuestas verdaderas estaban ocultas durante el desarrollo. En estas pruebas, su modelo igualó o superó de forma consistente métodos que combinan cálculos cuánticos intensivos con aprendizaje automático, así como otras redes neuronales basadas en grafos. De forma importante, también examinaron los casos donde las predicciones fallaron, rastreando a menudo las causas hasta sitios ionizables faltantes, tautómeros complicados que pueden redistribuir protones dentro de la molécula, o datos experimentales que eran en sí mismos ambiguos. En general, solo una pequeña fracción de compuestos mostró errores grandes, y el rendimiento se mantuvo estable a través de muchos grupos funcionales diferentes.
Qué significa esto para los químicos y más allá
El estudio muestra que las máquinas pueden beneficiarse de ser enseñadas como estudiantes. Al incorporar ideas químicas simples pero potentes directamente en un algoritmo de aprendizaje, los autores crearon un predictor de pKa que es a la vez preciso y más interpretable que los modelos de caja negra. Para los usuarios prácticos, esto supone estimaciones más rápidas y fiables de acidez y basicidad en una amplia gama de moléculas, ayudando a guiar el diseño de fármacos y otros esfuerzos de descubrimiento molecular. Más ampliamente, el trabajo sugiere un camino para herramientas futuras: en lugar de pedir a la inteligencia artificial que redescubra la ciencia fundamental desde cero, podemos integrar la experiencia humana y dejar que los modelos se centren en refinar y ampliar ese conocimiento.
Cita: Genzling, J., Luo, Z., Weiser, B. et al. Development of a pKa predictor (pKaLearn) by leveraging teaching experience to improve machine learning. Commun Chem 9, 181 (2026). https://doi.org/10.1038/s42004-026-01983-y
Palabras clave: predicción de pKa, aprendizaje automático, red neuronal gráfica, química computacional, diseño de fármacos