Clear Sky Science · it
Sviluppo di un predittore di pKa (pKaLearn) sfruttando l’esperienza didattica per migliorare il machine learning
Perché è importante insegnare alle macchine gli acidi
Prodotti di uso quotidiano, dai farmaci alle batterie, dipendono da quanto facilmente le molecole cedono o accettano protoni, una proprietà espressa dalla familiare scala del pH e dal suo cugino molecolare, il pKa. Misurare il pKa in laboratorio richiede tempo e lavoro, e persino metodi computazionali avanzati possono essere lenti o inaffidabili. Questo studio pone una domanda semplice ma dalle grandi implicazioni: cosa succederebbe se insegnassimo ai computer la chimica di base come facciamo con gli studenti, e poi chiedessimo loro di prevedere i valori di pKa più velocemente e con maggiore precisione?
Dalle regole della classe alle regole del computer
Nelle lezioni di chimica gli studenti imparano passo dopo passo. Prima vengono gli atomi e tendenze semplici come l’elettronegatività, poi concetti come risonanza, tensione degli anelli e come i gruppi vicini attirano o spingono gli elettroni. Solo più tardi affrontano previsioni difficili, come quale idrogeno in una molecola complessa sarà il più acido. Gli autori sostengono che la maggior parte dei modelli di machine learning salta questa progressione. Spesso ricevono in input strutture grezze o impronte astratte e devono scoprire i modelli da soli, il che può portarli a memorizzare esempi invece di comprendere le cause sottostanti. Qui, il gruppo codifica deliberatamente le stesse idee di base che usano gli insegnanti umani e le inserisce nei loro algoritmi.
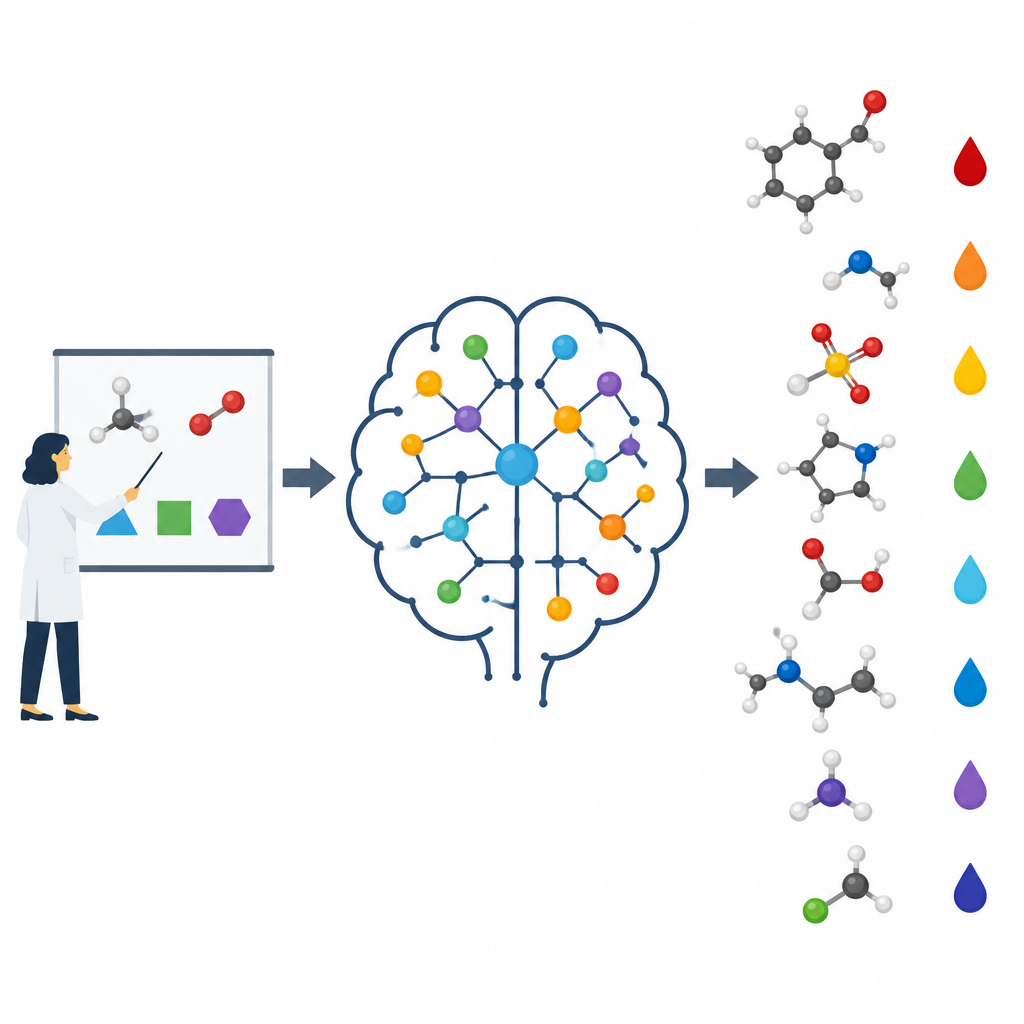
Costruire un modello che pensa come un chimico
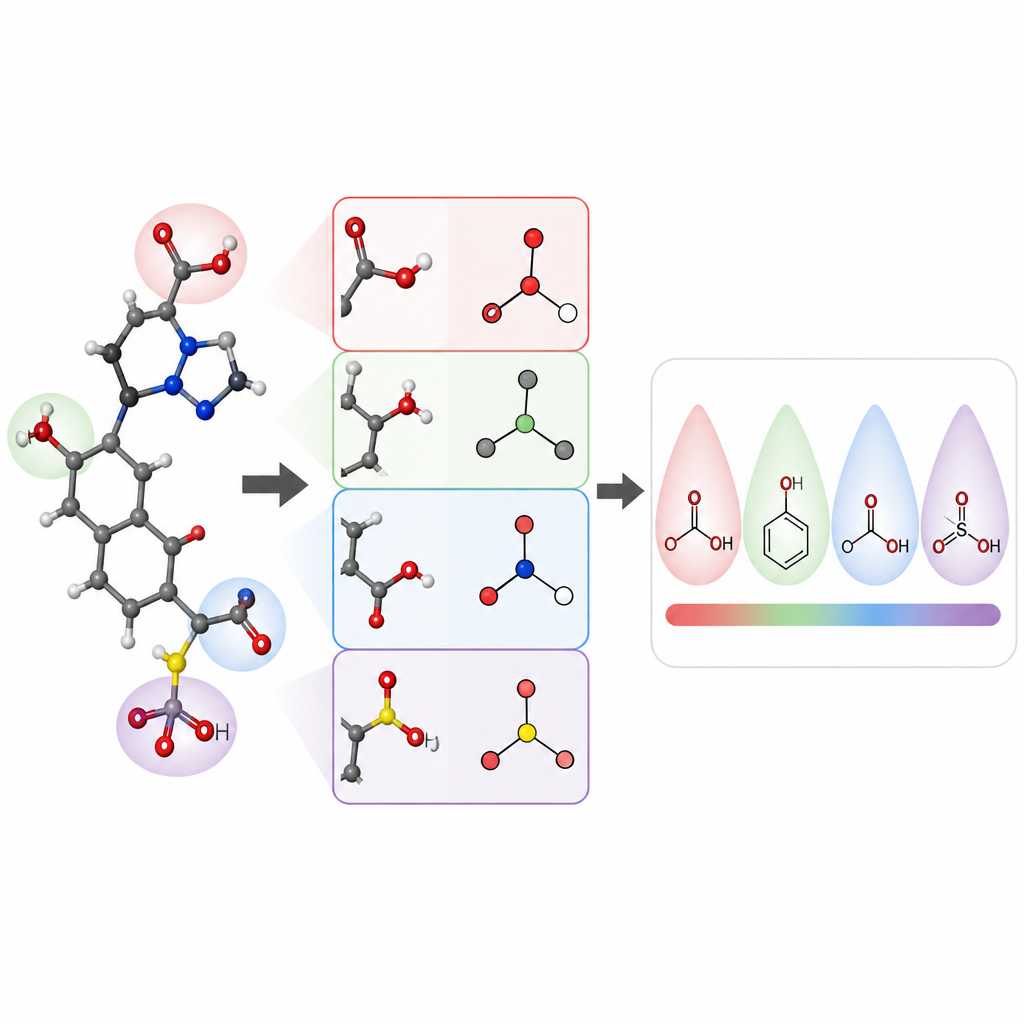
I ricercatori si sono concentrati sulla previsione dei valori di pKa di piccole molecole organiche, una proprietà centrale per la progettazione di farmaci e molte aree della chimica. Hanno costruito un modello chiamato pKaLearn utilizzando una rete neurale a grafo, che tratta una molecola come un insieme di atomi connessi da legami. Invece di affidarsi solo a codici strutturali generici, hanno aggiunto caratteristiche direttamente legate ai principi dei manuali: quanto è polare un legame, quanti legami separano un gruppo carico da un sito ionizzabile, se un legame fa parte di un anello e come coniugazione e risonanza possono distribuire la carica. Hanno inoltre misurato quanto lontano effetti sottili come la polarizzazione induttiva si propagano lungo una catena di atomi e progettato la rete in modo che ogni sito ionizzabile “percepisse” il suo ambiente fino a circa sette legami di distanza.
Insegnare, testare ed evitare la semplice memorizzazione
Per verificare se il loro approccio didattico aiutasse davvero il computer a imparare, gli autori hanno assemblato e pulito con cura un dataset di circa 13.000 valori di pKa. Invece della solita divisione casuale, che può rendere molecole di addestramento e di test molto simili, hanno raggruppato le molecole in modo che il set di test contenesse nuove famiglie chimiche. Questo esame più difficile rivela se il modello ha imparato regole generali o si è limitato a memorizzare esempi. In queste condizioni, pKaLearn ha raggiunto errori tipici inferiori a 0,7 unità di pKa, migliori dei modelli statistici tradizionali basati su fingerprint e più performante di diversi predittori esistenti basati su machine learning e chimica quantistica. Quando hanno provato scorciatoie standard, come affidarsi solo ai tipi di elemento o a una definizione software comune di legami coniugati, le prestazioni sono diminuite, sottolineando il valore di caratteristiche chimicamente significative e ben definite.
Confronto con altri predittori avanzati
Il gruppo ha confrontato pKaLearn con noti strumenti di predizione del pKa su set di benchmark largamente usati, inclusi composti provenienti da aziende farmaceutiche e sfide di previsione in cieco dove le risposte vere erano nascoste durante lo sviluppo. In questi test, il loro modello ha costantemente eguagliato o superato metodi che combinano pesanti calcoli quantistici con machine learning, così come altre reti neurali basate su grafi. È importante che abbiano anche esaminato i casi in cui le predizioni fallivano, spesso risalendo a siti ionizzabili mancanti, tautomerie insidiose che possono spostare protoni all’interno di una molecola o dati sperimentali a loro volta ambigui. Nel complesso, solo una piccola frazione di composti ha mostrato errori grandi, e le prestazioni sono rimaste stabili attraverso molti gruppi funzionali diversi.
Cosa significa per i chimici e oltre
Lo studio mostra che le macchine possono beneficiare di un insegnamento simile a quello rivolto agli studenti. Incorporando idee chimiche semplici ma potenti direttamente in un algoritmo di apprendimento, gli autori hanno creato un predittore di pKa che è sia preciso sia più interpretabile rispetto a modelli scatola nera. Per gli utenti pratici, questo si traduce in stime più rapide e più affidabili di acidità e basicità su un’ampia gamma di molecole, aiutando a guidare la progettazione di farmaci e altri sforzi di scoperta molecolare. Più in generale, il lavoro suggerisce una strada per strumenti futuri: invece di chiedere all’intelligenza artificiale di riscoprire la scienza fondamentale da zero, possiamo incorporare l’expertise umana e lasciare che i modelli si concentrino sul perfezionare ed estendere quella conoscenza.
Citazione: Genzling, J., Luo, Z., Weiser, B. et al. Development of a pKa predictor (pKaLearn) by leveraging teaching experience to improve machine learning. Commun Chem 9, 181 (2026). https://doi.org/10.1038/s42004-026-01983-y
Parole chiave: predizione pKa, machine learning, rete neurale a grafo, chimica computazionale, progettazione di farmaci