Clear Sky Science · sv
Att reda ut koevolutionära begränsningar för att modellera proteiners konformationella heterogenitet
Proteiner som formskiftande maskiner
Proteiner är de små maskiner som gör livet möjligt, och många av dem fungerar genom att subtilt byta form. Dessa skift kan slå på eller av signaler, öppna eller stänga molekylära portar eller omforma bindningsfickor som läkemedel försöker träffa. Ändå försöker de flesta datorverktyg fortfarande tilldela varje protein bara en "korrekt" struktur, vilket döljer den flexibilitet som ligger bakom både hälsa och sjukdom. I den här artikeln presenteras EvoSplit, ett nytt sätt att läsa den evolutionära information som är inbäddad i proteinsekvenser för att avslöja flera funktionellt viktiga former — inklusive sådana som aldrig setts i experiment och som kan öppna nya vägar för läkemedelsforskning.
Varför proteiners flexibilitet spelar roll för medicin
Inne i våra celler sitter proteiner sällan stilla. De böjer sig, vrider sig och ibland också omvandlar delar av sig själva som svar på förändringar i temperatur, surhetsgrad, bindningspartners eller kemiska modifieringar. Sådana rörelser kan vara små, som några sidokedjors förflyttning, eller dramatiska, som hela sektioner som vänder från en helix till ett ark. Dessa förändringar är centrala för hur receptorer känner av hormoner, hur transportörer flyttar molekyler över membraner och hur onkoproteiner driver cancer. Om vi bara känner till en ögonblicksbild av ett protein kan vi missa den aktiva formen, den läkemedelsbara fickan eller den konformation som leder till sjukdom. Att få en korrekt "rollista" över ett proteins stabila former är därför avgörande för att förstå biologin och utforma riktade terapier.
Att läsa evolutionens dolda anteckningar
Under miljontals år har proteinsekvenser utvecklats under tryck att bevara inte bara en enda struktur utan ofta flera biologiskt relevanta former. När två aminosyror tenderar att mutera koordinerat över närbesläktade proteiner antyder det att de måste förbli i kontakt i åtminstone en konformation. Moderna djupinlärningssystem som AlphaFold2 utmärker sig eftersom de utvinner sådana koevolutionära mönster från stora familjer av besläktade sekvenser, så kallade multiple sequence alignments. Men när ett protein kan anta mer än ett fold blir signalerna för olika tillstånd blandade, och standardmetoder tenderar oftast att kollapsa dem till en enda, genomsnittlig struktur. Tidigare metoder försökte skilja dessa åt genom att klustra sekvenser baserat på deras övergripande likhet, men sådana angreppssätt förbises i stor utsträckning de parvisa mönstren som faktiskt kodar för strukturella preferenser.
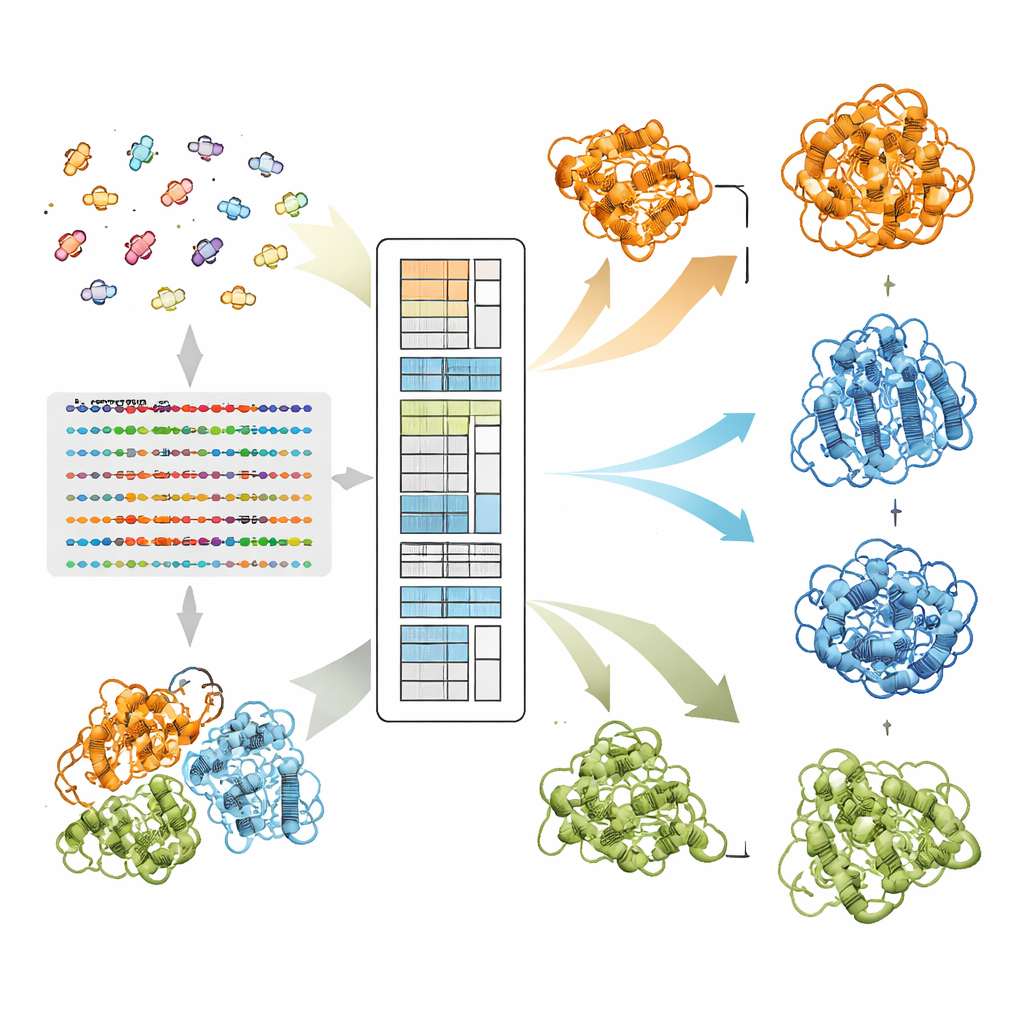
Hur EvoSplit separerar överlappande former
Författarna bygger vidare på en protein-språkmodell kallad MSA Transformer, som använder en attention-mekanism för att lära sig vilka rester i vilka sekvenser som "uppmärksammar" varandra. De visar att för proteiner med flera kända strukturer tenderar attention-mönstret för varje enskild sekvens att efterlikna kontaktkartan för en specifik konformation mer än för en annan. Med andra ord bär varje sekvens ett fingeravtryck av sin favoritform. EvoSplit utnyttjar detta genom att använda attention-matriserna — inte rå sekvenslikhet — som egenskaper för att klustra alignmenten i undergrupper. Varje kluster matas sedan separat in i AlphaFold2, vilket effektivt ger strukturprediktorn en renare, konformationsspecifik evolutionär prompt. Över 85 proteiner som är kända för att växla fold ger EvoSplit modeller som bättre överensstämmer med experimentella strukturer och med högre förtroende än en ledande sekvensbaserad klustringsmetod, särskilt för det mer sällan sampade tillståndet.
Att hitta nya tillstånd bortom träningsdata
En huvudfråga med kraftfulla neurala nätverk är att de kanske helt enkelt "minns" strukturer från sina träningsuppsättningar snarare än att upptäcka nya. För att testa om EvoSplit verkligen tillför information vänder sig författarna till en uppsättning transportörer och receptorer där alternativa tillstånd inte ingick i AlphaFold2:s ursprungliga träning. Även här återfinner EvoSplit både inåt- och utåtriktade former, liksom distinkta aktiva och inaktiva konformationer, med hög strukturell likhet mot experimentella modeller. Metoden skalar också till mer explorativa uppgifter: tillämpad på över hundra proteiner kopplade till mänsklig cancer flaggar den 54 kandidater som sannolikt antar flera konformationer. För några, såsom kinasen LCK och cellcykelregulatorn cyklin D1, föreslår EvoSplit trovärdiga domänarrangemang som ekar kända strukturer från besläktade proteiner, vilket antyder icke-observerade men biofysiskt rimliga tillstånd.
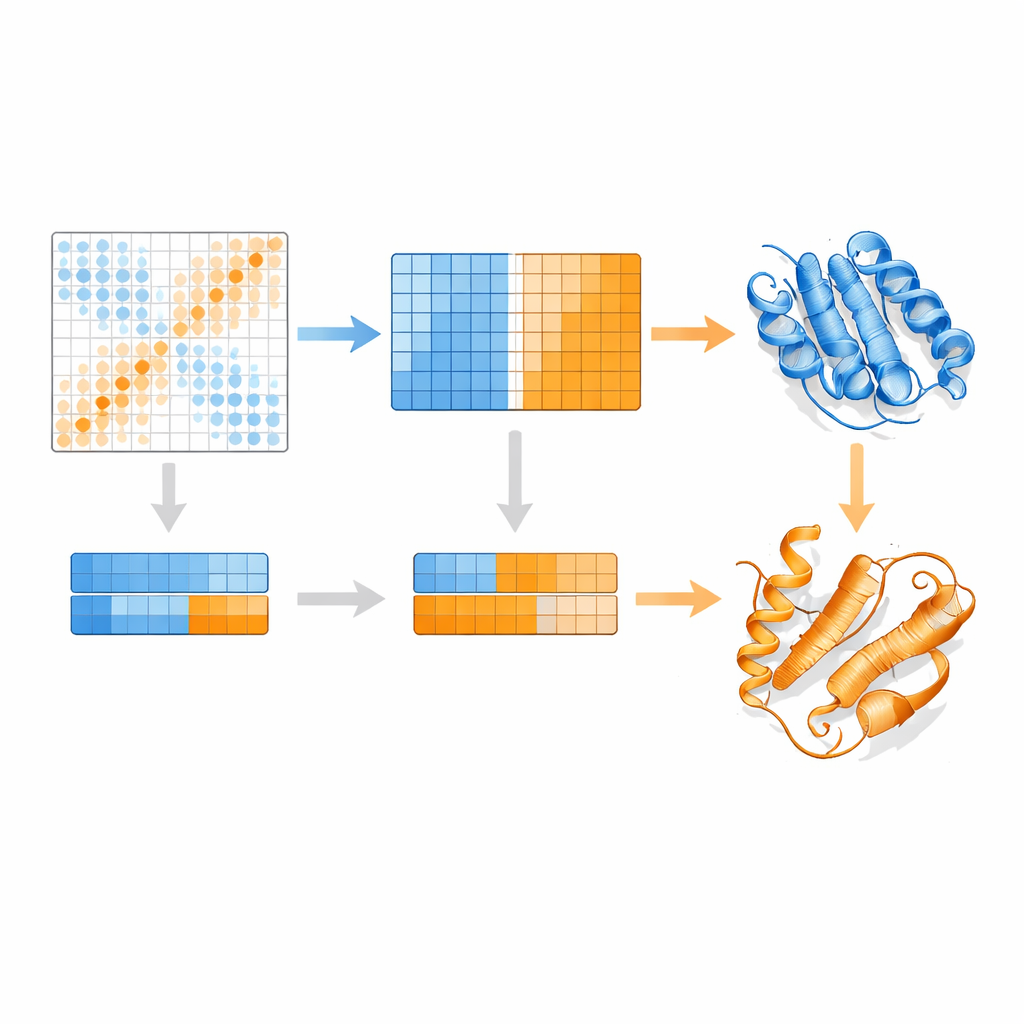
En överraskande ny fold i cancerkopplade switchar
Kanske är det mest fascinerande resultatet kopplat till små GTPaser som HRAS och KRAS, klassiska molekylära switchar som ofta muteras i tumörer. Dessa proteiner växlar normalt mellan "på" och "av" genom subtila omarrangemang nära nukleotidbindningsstället samtidigt som resten av deras fold förblir intakt. EvoSplit förutspår dock upprepade gånger en alternativ konformation där en helix nära proteinets början omvandlas till ett ark och därigenom ändrar den övergripande topologin. Detta mönster framträder över fem besläktade GTPaser, vilket tyder på att det inte är en slump. Simulationer av detta ovanliga tillstånd förblir stabila över hundratals nanosekunder, och analyser av evolutionära kopplingar visar distinkta kontaktsignaler som stämmer överens med dess unika ark-kontakter. När författarna modellerar interaktioner mellan HRAS och flera kända partners bildar både den klassiska och den nya konformationen stabila komplex, men med förskjutna kontaktgränssnitt, vilket antyder att den alternativa folden kan stödja olika signaleringsbeteenden.
Vad detta betyder för framtida läkemedelsdesign
För en icke-specialist är huvudbudskapet att våra proteiner kan dölja fler former — och därmed fler funktionella möjligheter — än vad traditionell strukturprediktion har visat. EvoSplit använder evolutionstyrd mönsterigenkänning för att separera dessa dolda tillstånd i stället för att jämna ut dem. Genom att överträffa tidigare metoder på kända fold-växlande proteiner, upptäcka alternativa tillstånd i välstuderade receptorer och transportörer och föreslå en ny, stabil fold för cancerrelaterade switchar som HRAS, argumenterar detta arbete för att modellering av flera tillstånd bör bli rutin. I praktiska termer kan rikare strukturkataloger lyfta fram nya fickor för läkemedel, förklara varför vissa mutationer är skadliga och peka ut vägar som bara blir synliga när vi ser bortom en enda statisk struktur.
Citering: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Nyckelord: proteiners konformationella dynamik, koevolutionära signaler, AlphaFold2, cancerrelaterade proteiner, GTPas vikningsbyte