Clear Sky Science · fr
Démêler les contraintes de coévolution pour modéliser l'hétérogénéité conformationnelle des protéines
Les protéines, machines changeant de forme
Les protéines sont les petites machines qui rendent la vie possible, et nombre d'entre elles fonctionnent en modifiant subtilement leur forme. Ces changements peuvent activer ou désactiver des signaux, ouvrir ou fermer des portes moléculaires, ou remodeler des poches de liaison ciblées par des médicaments. Pourtant, la plupart des outils informatiques cherchent encore à attribuer à chaque protéine une seule « structure correcte », masquant la flexibilité même qui sous-tend la santé et la maladie. Cet article présente EvoSplit, une nouvelle méthode pour lire l'empreinte évolutive inscrite dans les séquences protéiques afin de révéler plusieurs formes fonctionnellement importantes — y compris certaines jamais observées expérimentalement et susceptibles d'ouvrir de nouvelles voies pour la découverte de médicaments.
Pourquoi la flexibilité des protéines compte pour la médecine
À l'intérieur de nos cellules, les protéines sont rarement immobiles. Elles se plient, se tordent et refoldent parfois des portions d'elles-mêmes en réponse à des variations de température, d'acidité, de partenaires de liaison ou de modifications chimiques. Ces mouvements peuvent être modestes, comme le déplacement de quelques chaînes latérales, ou spectaculaires, comme la transformation complète d'une hélice en feuillet. Ces changements sont au cœur du fonctionnement des récepteurs sensibles aux hormones, des transporteurs qui déplacent des molécules à travers les membranes, et des oncoprotéines qui pilotent le cancer. Si l'on ne connaît qu'un seul instantané d'une protéine, on peut manquer la forme active, la poche exploitable par un médicament ou la conformation conduisant à la maladie. Dresser une « liste complète » des formes stables d'une protéine est donc crucial pour comprendre la biologie et concevoir des thérapies ciblées.
Lire les notes cachées de l'évolution
Sur des millions d'années, les séquences protéiques ont évolué sous pression pour préserver non seulement une structure unique, mais souvent plusieurs formes biologiquement pertinentes. Lorsque deux acides aminés ont tendance à muter de façon coordonnée à travers des protéines apparentées, cela suggère qu'ils doivent rester en contact dans au moins une conformation. Les systèmes modernes d'apprentissage profond comme AlphaFold2 excellent parce qu'ils exploitent ces schémas de coévolution à partir de vastes familles de séquences apparentées, appelées alignements multiples de séquences. Cependant, lorsqu'une protéine peut adopter plusieurs repliements, les signaux correspondant aux différents états se mêlent, et les approches standards les condensent généralement en une seule structure moyenne. Des méthodes antérieures ont tenté de démêler cela en regroupant les séquences selon leur similarité globale, mais ces approches ont largement ignoré les schémas pair-à-pair qui encodent réellement les préférences structurales.
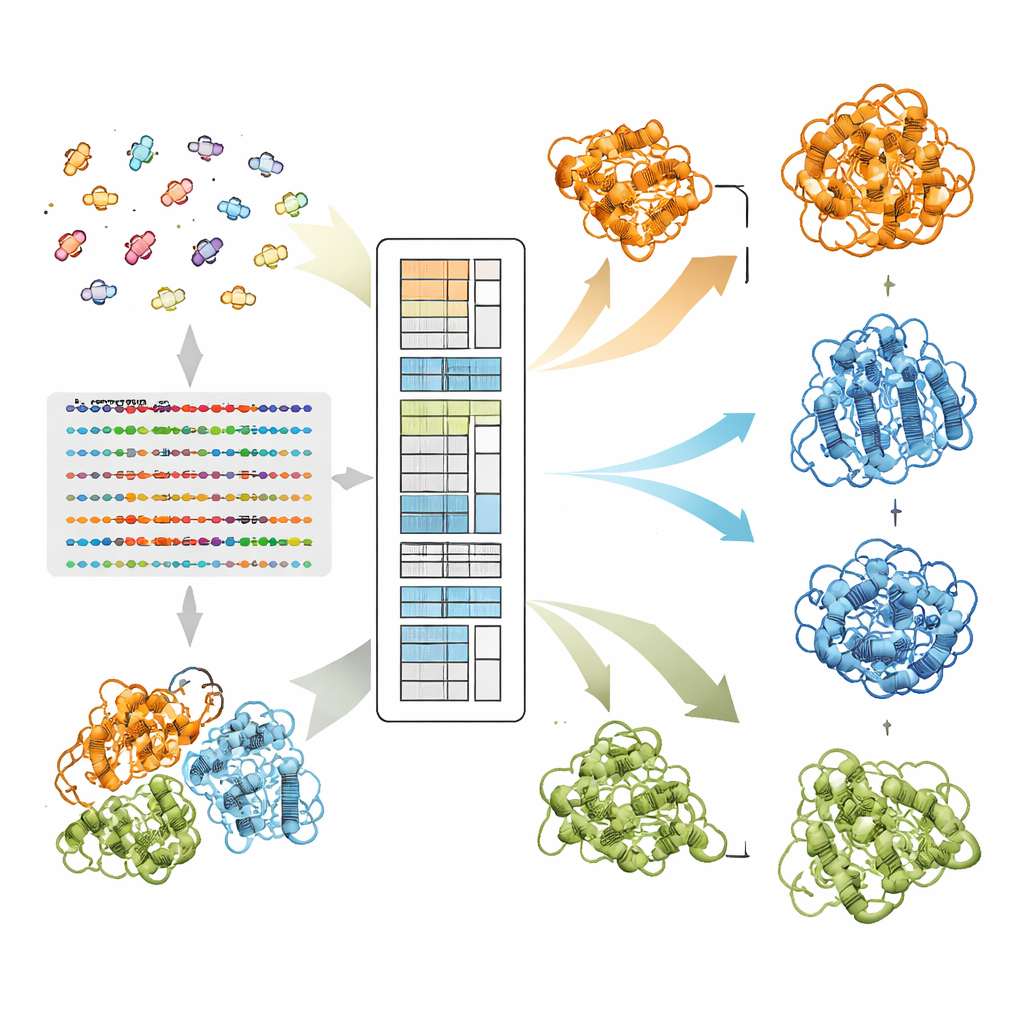
Comment EvoSplit sépare les formes qui se chevauchent
Les auteurs s'appuient sur un modèle langage protéique appelé MSA Transformer, qui utilise un mécanisme d'attention pour apprendre quelles résidus dans quelles séquences « se regardent ». Ils montrent que, pour les protéines ayant plusieurs structures connues, le motif d'attention de chaque séquence individuelle tend à ressembler davantage à la carte de contacts d'une conformation spécifique qu'à une autre. Autrement dit, chaque séquence porte l'empreinte de sa forme préférée. EvoSplit exploite cela en utilisant les matrices d'attention — et non la similarité brute des séquences — comme caractéristiques pour regrouper l'alignement en sous-groupes. Chaque cluster est ensuite soumis séparément à AlphaFold2, donnant au prédicteur de structures une impulsion évolutive plus propre et spécifique à une conformation. Sur 85 protéines connues pour changer de repliement, EvoSplit produit des modèles qui concordent mieux avec les structures expérimentales et avec une confiance plus élevée que la principale méthode de clustering basée sur les séquences, en particulier pour l'état le moins fréquemment échantillonné.
Découvrir des états nouveaux au-delà des données d'entraînement
Un sujet de préoccupation avec les réseaux neuronaux puissants est qu'ils puissent simplement « mémoriser » des structures issues de leurs jeux d'entraînement plutôt que d'en découvrir de nouvelles. Pour vérifier si EvoSplit apporte réellement de l'information, les auteurs se tournent vers un ensemble de transporteurs et de récepteurs dont les états alternatifs n'étaient pas inclus dans l'entraînement initial d'AlphaFold2. Même dans ces cas, EvoSplit retrouve des formes orientées vers l'intérieur et l'extérieur, ainsi que des conformations actives et inactives distinctes, avec une forte similarité structurale aux modèles expérimentaux. La méthode s'étend aussi à des tâches plus exploratoires : appliquée à plus d'une centaine de protéines liées aux cancers humains, elle signale 54 candidats susceptibles d'adopter plusieurs conformations. Pour certaines, comme la kinase LCK et le régulateur du cycle cellulaire cycline D1, EvoSplit propose des arrangements plausibles de domaines rappelant des structures connues de protéines apparentées, suggérant des états non observés mais biophysiquement raisonnables.
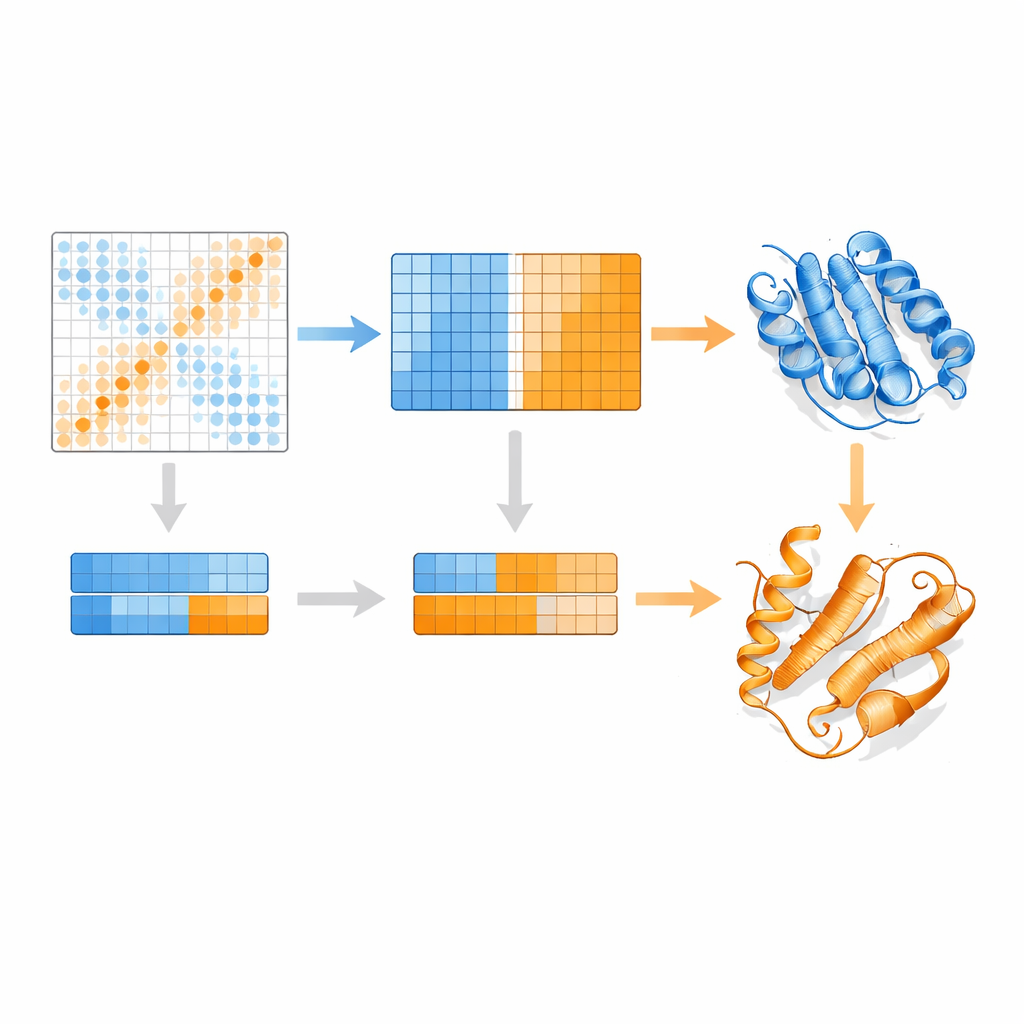
Un nouveau repliement surprenant dans des commutateurs liés au cancer
Peut-être le résultat le plus intrigant concerne de petites GTPases telles que HRAS et KRAS, commutateurs moléculaires classiques fréquemment mutés dans les tumeurs. Ces protéines basculent normalement entre « on » et « off » par de subtiles réarrangements près du site de liaison au nucléotide tout en conservant le reste de leur repliement. EvoSplit prédit cependant de façon récurrente une conformation alternative dans laquelle une hélice proche du début de la protéine se convertit en feuillet, modifiant la topologie globale. Ce motif apparaît chez cinq GTPases apparentées, ce qui suggère qu'il ne s'agit pas d'un artefact. Des simulations de cet état inhabituel restent stables sur plusieurs centaines de nanosecondes, et des analyses des couplages évolutifs montrent des signaux de contacts distincts qui correspondent à ses contacts en feuillet uniques. Lorsque les auteurs modélisent les interactions entre HRAS et plusieurs partenaires connus, la conformation classique et la nouvelle forment toutes deux des complexes stables, mais avec des interfaces de contact décalées, ce qui implique que le repliement alternatif pourrait soutenir des comportements de signalisation différents.
Ce que cela signifie pour la conception de médicaments à venir
Pour un non-spécialiste, le message central est que nos protéines peuvent abriter plus de formes — et donc plus de possibilités fonctionnelles — que ce que la prédiction de structures traditionnelle a révélé. EvoSplit utilise la reconnaissance de motifs guidée par l'évolution pour séparer ces états cachés au lieu de les moyenner. En dépassant les méthodes antérieures sur des protéines connues pour changer de repliement, en découvrant des états alternatifs dans des récepteurs et transporteurs bien étudiés, et en suggérant un nouveau repliement stable pour des commutateurs liés au cancer comme HRAS, ce travail plaide pour que la modélisation multi-états devienne la norme. Concrètement, des catalogues structuraux plus riches pourraient mettre en évidence de nouvelles poches pour les médicaments, expliquer pourquoi certaines mutations sont délétères, et révéler des voies fonctionnelles qui n'apparaissent que lorsqu'on dépasse la vision d'une structure statique unique.
Citation: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Mots-clés: dynamique conformationnelle des protéines, signaux de coévolution, AlphaFold2, protéines liées au cancer, changement de repliement GTPase