Clear Sky Science · it
Disinnescare i vincoli coevolutivi per modellare l’eterogeneità conformazionale delle proteine
Proteine come macchine che cambiano forma
Le proteine sono le piccole macchine che rendono possibile la vita, e molte di esse funzionano cambiando forma in modo sottile. Questi spostamenti possono attivare o disattivare segnali, aprire o chiudere porte molecolari, o rimodellare tasche di legame su cui i farmaci mirano. Eppure la maggior parte degli strumenti informatici continua a cercare di assegnare a ciascuna proteina una sola struttura “corretta”, nascondendo la flessibilità che è alla base della salute e della malattia. Questo articolo presenta EvoSplit, un nuovo modo di leggere il documento evolutivo codificato nelle sequenze proteiche per scoprire più forme funzionalmente importanti — incluse alcune mai osservate sperimentalmente e che potrebbero aprire nuove strade per la scoperta di farmaci.
Perché la flessibilità delle proteine è importante per la medicina
All’interno delle nostre cellule, le proteine raramente stanno ferme. Si piegano, si torcono e talvolta riorganizzano parti di sé in risposta a cambi di temperatura, acidità, partner di legame o modifiche chimiche. Questi movimenti possono essere piccoli, come lo spostamento di alcune catene laterali, o drammatici, come intere porzioni che passano da un’elica a un foglietto. Tali cambiamenti sono centrali per il modo in cui i recettori percepiscono gli ormoni, come i trasportatori muovono molecole attraverso le membrane e come le oncoproteine guidano il cancro. Se conosciamo solo un’istantanea di una proteina, potremmo perdere la forma attiva, la tasca sfruttabile dai farmaci o la conformazione che porta alla malattia. Catturare un elenco accurato delle forme stabili di una proteina è quindi cruciale per comprendere la biologia e progettare terapie mirate.
Leggere le note nascoste dell’evoluzione
Nel corso di milioni di anni, le sequenze proteiche si sono evolute sotto la pressione di preservare non solo una singola struttura, ma spesso diverse forme rilevanti biologicamente. Quando due amminoacidi tendono a mutare in modo coordinato attraverso proteine correlate, questo suggerisce che devono rimanere a contatto in almeno una conformazione. I moderni sistemi di deep learning come AlphaFold2 eccellono perché estraggono tali schemi coevolutivi da vaste famiglie di sequenze correlate, note come allineamenti di sequenze multiple. Tuttavia, quando una proteina può assumere più di un fold, i segnali per diversi stati si mescolano e gli approcci standard di solito li comprimono in una singola struttura mediata. Metodi precedenti hanno tentato di separare questi segnali raggruppando le sequenze in base alla somiglianza complessiva, ma quegli approcci in gran parte hanno ignorato i pattern a coppie che effettivamente codificano le preferenze strutturali.
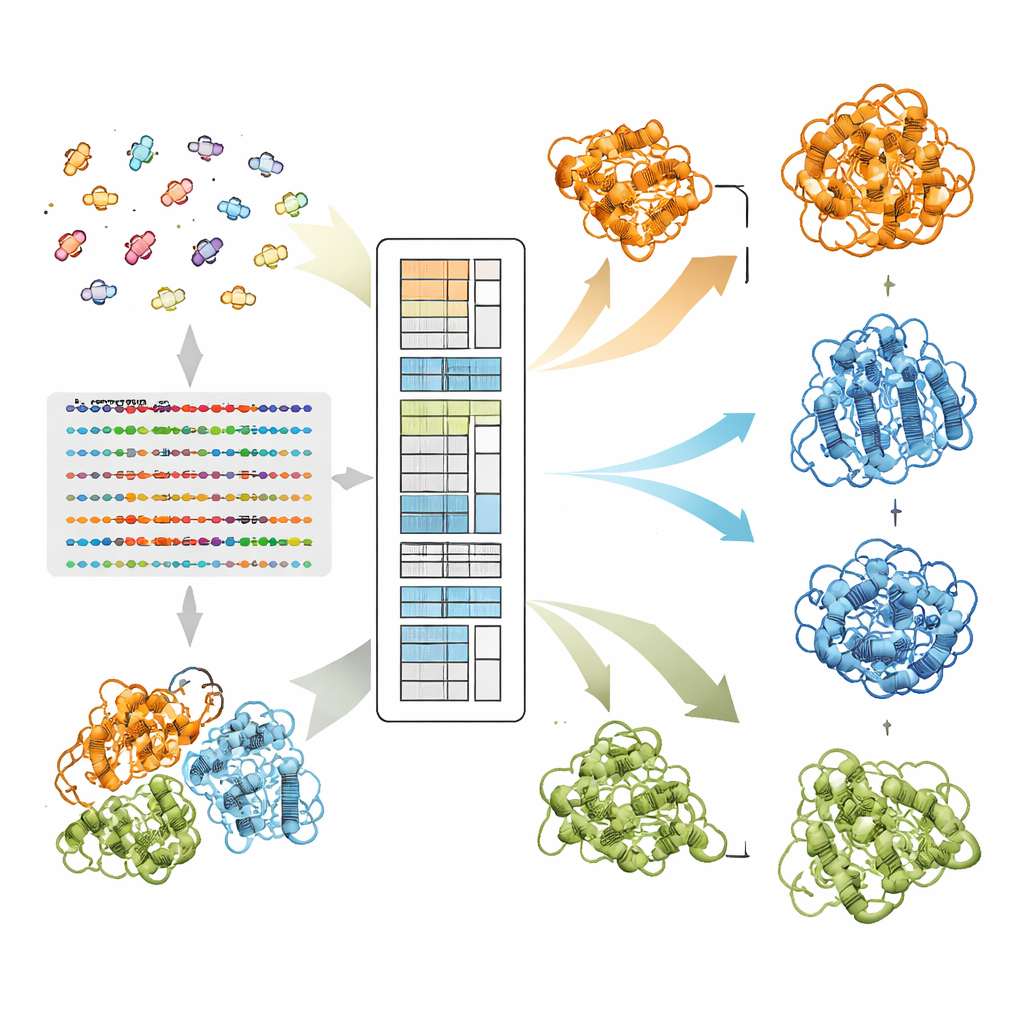
Come EvoSplit separa forme sovrapposte
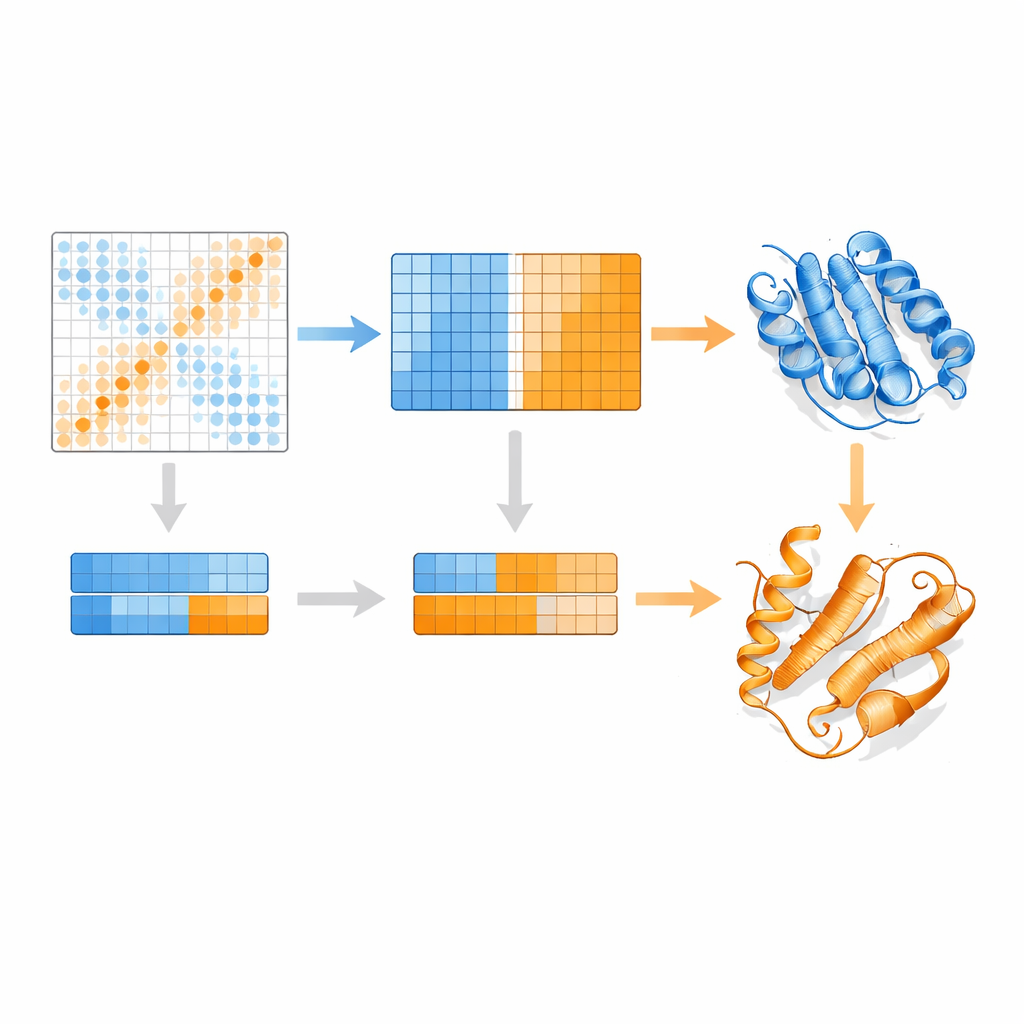
Gli autori si basano su un modello linguistico per proteine chiamato MSA Transformer, che usa un meccanismo di attenzione per apprendere quali residui in quali sequenze “prestano attenzione” l’uno all’altro. Mostrano che, per proteine con più strutture note, il pattern di attenzione di ogni singola sequenza tende a somigliare alla mappa di contatti di una specifica conformazione più che all’altra. In altre parole, ogni sequenza porta l’impronta della sua forma preferita. EvoSplit sfrutta questo usando le matrici di attenzione — non la somiglianza grezza delle sequenze — come caratteristiche per raggruppare l’allineamento in sottogruppi. Ogni cluster viene poi fornito separatamente ad AlphaFold2, dando al predittore strutturale un prompt evolutivo più pulito e specifico per la conformazione. Su 85 proteine note per cambiare fold, EvoSplit produce modelli che concordano meglio con le strutture sperimentali e con maggiore confidenza rispetto a un metodo di clustering basato sulle sequenze leader, soprattutto per lo stato più raramente campionato.
Trovare nuovi stati oltre i dati di addestramento
Una preoccupazione chiave con potenti reti neurali è che possano semplicemente “ricordare” strutture dai loro set di addestramento invece di scoprirne di nuove. Per verificare se EvoSplit aggiunge veramente informazione, gli autori esaminano un insieme di trasportatori e recettori i cui stati alternativi non erano inclusi nell’addestramento originale di AlphaFold2. Anche qui, EvoSplit recupera sia le forme rivolte verso l’interno sia quelle verso l’esterno, così come forme attive e inattive distinte, con alta somiglianza strutturale ai modelli sperimentali. Il metodo scala anche a compiti più esplorativi: applicato a oltre cento proteine collegate ai tumori umani, individua 54 candidati probabilmente in grado di adottare più conformazioni. Per alcuni, come la chinasi LCK e il regolatore del ciclo cellulare ciclina D1, EvoSplit suggerisce arrangiamenti plausibili dei domini che richiamano strutture note di proteine correlate, suggerendo stati non osservati ma biofisicamente ragionevoli.
Un nuovo fold sorprendente negli switch legati al cancro
Forse il risultato più intrigante riguarda piccole GTPasi come HRAS e KRAS, classici interruttori molecolari frequentemente mutati nei tumori. Queste proteine normalmente oscillano tra “on” e “off” mediante riorganizzazioni sottili vicino al sito di legame del nucleotide mantenendo intatto il resto del fold. EvoSplit, tuttavia, predice ripetutamente una conformazione alternativa in cui un’elica vicino all’inizio della proteina si converte in un foglietto, alterando la topologia complessiva. Questo pattern appare in cinque GTPasi correlate, suggerendo che non sia un caso. Simulazioni di questo stato insolito rimangono stabili per centinaia di nanosecondi, e analisi dei couplings evolutivi mostrano segnali di contatto distinti che corrispondono ai suoi contatti a foglietto unici. Quando gli autori modellano le interazioni tra HRAS e diversi partner noti, sia la conformazione classica sia quella nuova formano complessi stabili, ma con interfacce di contatto spostate, implicando che il fold alternativo potrebbe supportare comportamenti di segnalazione differenti.
Che cosa significa per il futuro della progettazione di farmaci
Per un non specialista, il messaggio principale è che le nostre proteine possono nascondere più forme — e quindi più possibilità funzionali — di quanto la predizione strutturale tradizionale abbia rivelato. EvoSplit usa il riconoscimento di pattern guidato dall’evoluzione per separare questi stati nascosti invece di mediare su di essi. Superando metodi precedenti su proteine note per il cambio di fold, scoprendo stati alternativi in recettori e trasportatori ben studiati e suggerendo un nuovo fold stabile per switch legati al cancro come HRAS, questo lavoro sostiene che la modellazione multi-stato dovrebbe diventare la norma. In termini pratici, cataloghi strutturali più ricchi potrebbero evidenziare nuove tasche per farmaci, spiegare perché certe mutazioni sono dannose e indicare vie che emergono solo quando si guarda oltre una singola struttura statica.
Citazione: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Parole chiave: dinamica conformazionale delle proteine, segnali coevolutivi, AlphaFold2, proteine correlate al cancro, switching del fold GTPasi