Clear Sky Science · pt
Desvendando restrições coevolutivas para modelar a heterogeneidade conformacional de proteínas
Proteínas como máquinas que mudam de forma
Proteínas são as pequenas máquinas que tornam a vida possível, e muitas funcionam ao alterar sua forma de maneira sutil. Essas mudanças podem ativar ou desativar sinais, abrir ou fechar portões moleculares, ou remodelar bolsões de ligação que fármacos procuram atingir. Ainda assim, a maioria das ferramentas computacionais tenta atribuir a cada proteína apenas uma estrutura “correta”, ocultando a flexibilidade que fundamenta saúde e doença. Este artigo apresenta o EvoSplit, uma nova forma de ler o registro evolutivo codificado em sequências de proteínas para revelar múltiplas formas funcionalmente importantes — incluindo algumas ainda não observadas experimentalmente e que podem abrir novas avenidas para descoberta de fármacos.
Por que a flexibilidade proteica importa para a medicina
Dentro de nossas células, proteínas raramente ficam paradas. Elas dobram, torcem e às vezes até refazem partes de si em resposta a mudanças de temperatura, acidez, parceiros de ligação ou modificações químicas. Esses movimentos podem ser pequenos, como a deslocação de alguns grupos laterais, ou dramáticos, como seções inteiras que mudam de uma hélice para uma folha. Essas alterações são centrais para como receptores detectam hormônios, como transportadores movem moléculas através de membranas e como oncoproteínas promovem o câncer. Se conhecermos apenas um instantâneo de uma proteína, podemos perder a forma ativa, o bolso passível de ser alvo de drogas ou a conformação que leva à doença. Capturar uma “lista de elencos” precisa das formas estáveis de uma proteína é, portanto, crucial para entender a biologia e projetar terapias direcionadas.
Lendo as notas ocultas da evolução
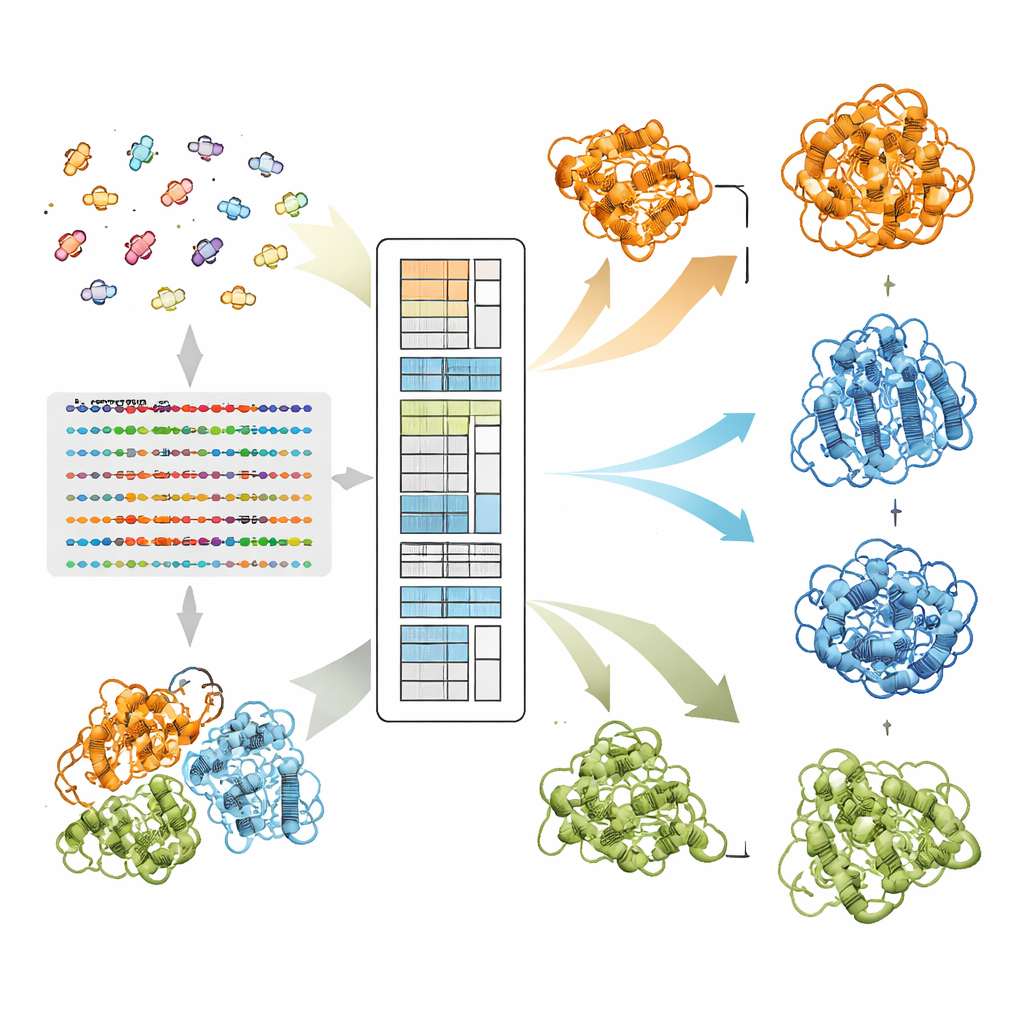
Ao longo de milhões de anos, sequências proteicas evoluíram sob pressão para preservar não apenas uma única estrutura, mas frequentemente várias formas biologicamente relevantes. Quando dois aminoácidos tendem a mutar de maneira coordenada entre proteínas relacionadas, isso sugere que eles devem permanecer em contato em pelo menos uma conformação. Sistemas modernos de aprendizado profundo como o AlphaFold2 se destacam porque extraem esses padrões coevolutivos de grandes famílias de sequências relacionadas, conhecidas como alinhamentos múltiplos de sequências. Entretanto, quando uma proteína pode adotar mais de uma dobra, os sinais para diferentes estados se misturam, e abordagens padrão tendem a colapsá-los em uma única estrutura média. Métodos anteriores tentaram separar isso agrupando sequências com base na similaridade global, mas essas abordagens em grande parte ignoraram os padrões pareados que de fato codificam preferências estruturais.
Como o EvoSplit separa formas sobrepostas
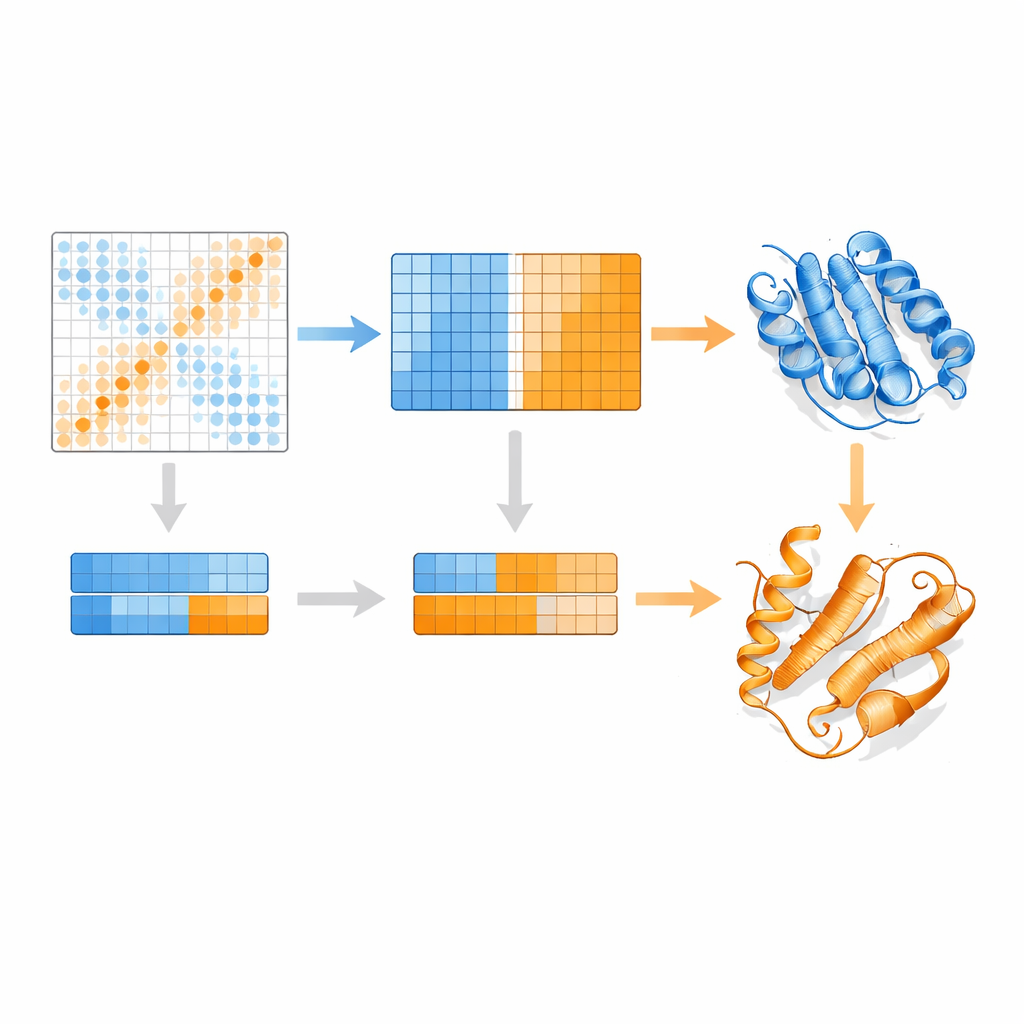
Os autores partem de um modelo de linguagem para proteínas chamado MSA Transformer, que usa um mecanismo de atenção para aprender quais resíduos em quais sequências “prestam atenção” uns aos outros. Eles mostram que, para proteínas com múltiplas estruturas conhecidas, o padrão de atenção de cada sequência individual tende a se assemelhar ao mapa de contatos de uma conformação específica mais do que a outra. Em outras palavras, cada sequência carrega uma impressão digital de sua forma preferida. O EvoSplit explora isso usando as matrizes de atenção — não a similaridade bruta de sequências — como características para agrupar o alinhamento em subgrupos. Cada cluster é então fornecido separadamente ao AlphaFold2, dando ao preditor de estrutura um estímulo evolutivo mais limpo e específico por conformação. Em 85 proteínas conhecidas por trocar de dobra, o EvoSplit produz modelos que concordam melhor com estruturas experimentais e com maior confiança do que um método de clustering baseado em sequência líder, especialmente para o estado menos amostrado.
Encontrando novos estados além dos dados de treinamento
Uma preocupação central com redes neurais poderosas é que elas possam simplesmente “memorizar” estruturas dos conjuntos de treinamento em vez de descobrir novas. Para testar se o EvoSplit realmente acrescenta informação, os autores analisam um conjunto de transportadores e receptores cujos estados alternativos não foram incluídos no treinamento original do AlphaFold2. Mesmo nesses casos, o EvoSplit recupera formas voltadas para o interior e para o exterior, bem como formas ativas e inativas distintas, com alta similaridade estrutural aos modelos experimentais. O método também escala para tarefas mais exploratórias: aplicado a mais de uma centena de proteínas ligadas ao câncer humano, assinala 54 candidatas que provavelmente adotam múltiplas conformações. Para algumas, como a quinase LCK e o regulador do ciclo celular ciclina D1, o EvoSplit sugere arranjos plausíveis de domínios que ecoam estruturas conhecidas de proteínas relacionadas, apontando para estados não observados, mas bi fisicamente razoáveis.
Uma nova dobra surpreendente em chaves ligadas ao câncer
Talvez o resultado mais intrigante envolva pequenas GTPases como HRAS e KRAS, interruptores moleculares clássicos frequentemente mutados em tumores. Essas proteínas normalmente alternam entre “ligado” e “desligado” por rearranjos sutis perto do sítio de ligação ao nucleotídeo, mantendo o restante da dobra intacto. O EvoSplit, porém, prevê repetidamente uma conformação alternativa em que uma hélice próxima ao início da proteína se converte em uma folha, alterando a topologia geral. Esse padrão aparece em cinco GTPases relacionadas, sugerindo que não é um acaso. Simulações desse estado incomum permanecem estáveis por centenas de nanosegundos, e análises de acoplamentos evolutivos mostram sinais de contato distintos que se alinham com seus contatos em folha únicos. Quando os autores modelam interações entre HRAS e vários parceiros conhecidos, tanto a conformação clássica quanto a nova formam complexos estáveis, porém com interfaces de contato deslocadas, o que implica que a dobra alternativa poderia sustentar comportamentos de sinalização diferentes.
O que isso significa para o design de fármacos futuro
Para um não especialista, a mensagem central é que nossas proteínas podem abrigar mais formas — e, portanto, mais possibilidades funcionais — do que a predição estrutural tradicional revelou. O EvoSplit usa reconhecimento de padrões guiado pela evolução para separar esses estados ocultos em vez de diluí-los. Ao superar métodos anteriores em proteínas conhecidas por troca de dobra, descobrir estados alternativos em receptores e transportadores bem estudados e sugerir uma nova dobra estável para interruptores relacionados ao câncer como HRAS, este trabalho argumenta que a modelagem multi-estado deveria se tornar rotineira. Em termos práticos, catálogos estruturais mais ricos poderiam destacar novos bolsões para fármacos, explicar por que certas mutações são prejudiciais e apontar caminhos que só se tornam visíveis quando olhamos além de uma única estrutura estática.
Citação: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Palavras-chave: dinâmica conformacional de proteínas, sinais coevolutivos, AlphaFold2, proteínas relacionadas ao câncer, troca de dobra em GTPases