Clear Sky Science · es
Desentrañando las restricciones coevolutivas para modelar la heterogeneidad conformacional de las proteínas
Proteínas como máquinas que cambian de forma
Las proteínas son las pequeñas máquinas que permiten la vida, y muchas de ellas funcionan al cambiar de forma de manera sutil. Estos cambios pueden activar o desactivar señales, abrir o cerrar compuertas moleculares, o remodelar bolsillos de unión que los fármacos intentan alcanzar. Sin embargo, la mayoría de las herramientas informáticas todavía intentan asignar a cada proteína una única estructura «correcta», ocultando la flexibilidad que subyace a la salud y a la enfermedad. Este artículo presenta EvoSplit, una nueva forma de leer el registro evolutivo codificado en las secuencias de proteínas para descubrir múltiples formas relevantes funcionalmente, incluidas algunas que nunca se han observado experimentalmente y que podrían abrir nuevas vías para el descubrimiento de fármacos.
Por qué la flexibilidad de las proteínas importa en medicina
Dentro de nuestras células, las proteínas rara vez están inmóviles. Se doblan, giran y, a veces, incluso repliegan partes de sí mismas en respuesta a cambios de temperatura, acidez, socios de unión o modificaciones químicas. Esos movimientos pueden ser pequeños, como el desplazamiento de unas pocas cadenas laterales, o drásticos, como secciones enteras que pasan de una hélice a una lámina. Estos cambios son centrales para cómo los receptores detectan hormonas, cómo los transportadores desplazan moléculas a través de membranas y cómo las oncoproteínas impulsan el cáncer. Si solo conocemos una instantánea de una proteína, podemos perder la forma activa, el bolsillo susceptible de ser atacado por un fármaco o la conformación que conduce a la enfermedad. Capturar un «reparto» preciso de las formas estables de una proteína es, por tanto, crucial para entender la biología y diseñar terapias dirigidas.
Leyendo las notas ocultas de la evolución
A lo largo de millones de años, las secuencias de proteínas han evolucionado bajo la presión de conservar no solo una estructura única, sino a menudo varias formas relevantes biológicamente. Cuando dos aminoácidos tienden a mutar de manera coordinada en proteínas relacionadas, sugiere que deben permanecer en contacto en al menos una conformación. Los sistemas modernos de aprendizaje profundo, como AlphaFold2, destacan porque extraen estos patrones coevolutivos de grandes familias de secuencias relacionadas, conocidas como alineamientos de secuencias múltiples. Sin embargo, cuando una proteína puede adoptar más de un pliegue, las señales de los distintos estados se mezclan y los enfoques estándar suelen colapsarlas en una única estructura promediada. Métodos anteriores intentaron separar esto agrupando secuencias según su similitud global, pero esos enfoques en gran medida pasaban por alto los patrones pares-a-pares que realmente codifican las preferencias estructurales.
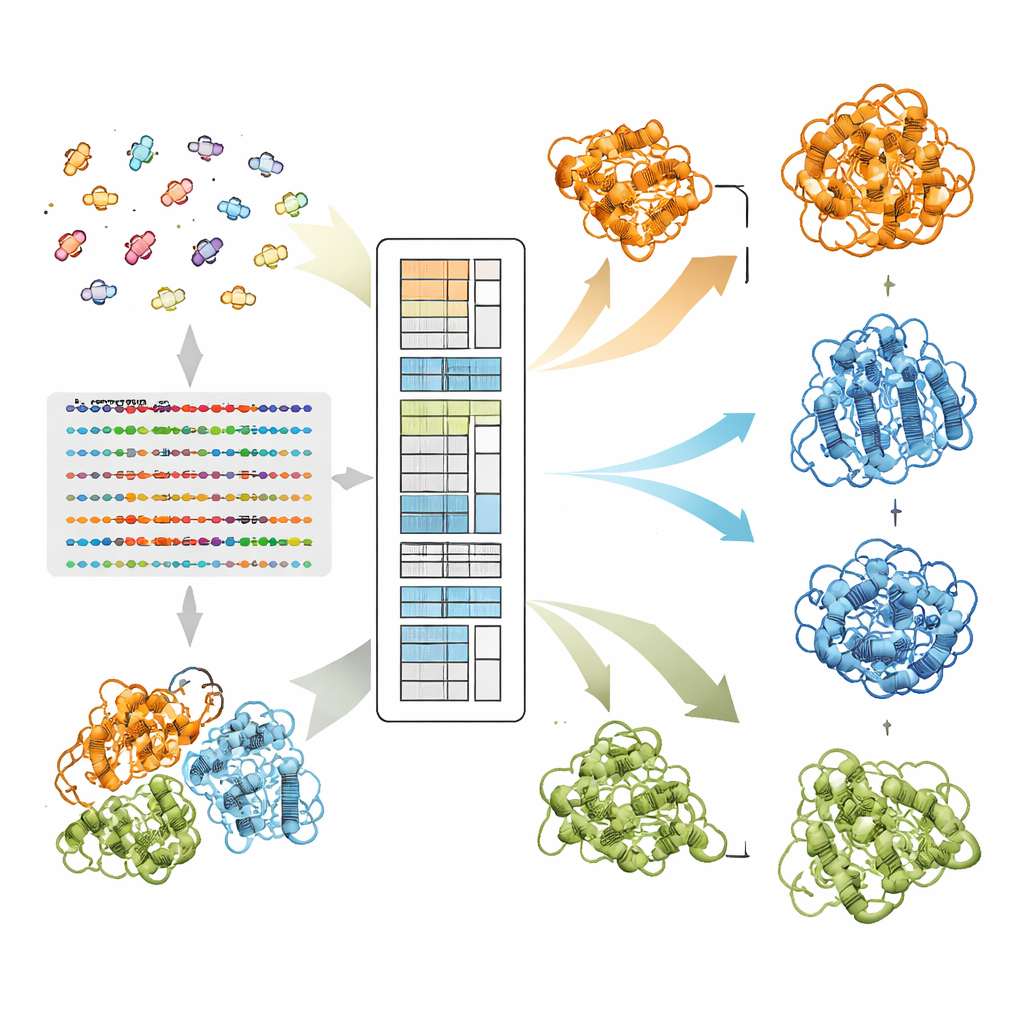
Cómo EvoSplit separa formas superpuestas
Los autores se basan en un modelo de lenguaje para proteínas llamado MSA Transformer, que utiliza un mecanismo de atención para aprender qué residuos en qué secuencias «prestan atención» entre sí. Demuestran que, para proteínas con múltiples estructuras conocidas, el patrón de atención de cada secuencia individual tiende a asemejarse más al mapa de contactos de una conformación específica que al de otra. En otras palabras, cada secuencia lleva la huella de su forma preferida. EvoSplit aprovecha esto usando las matrices de atención —no la similitud de secuencia cruda— como características para agrupar el alineamiento en subgrupos. Cada clúster se introduce por separado en AlphaFold2, dando al predictor de estructuras un estímulo evolutivo más limpio y específico de conformación. En 85 proteínas conocidas por cambiar de pliegue, EvoSplit produce modelos que concuerdan mejor con las estructuras experimentales y con mayor confianza que un método líder basado en el agrupamiento por secuencia, especialmente para el estado menos muestreado.
Encontrando nuevos estados más allá de los datos de entrenamiento
Una preocupación clave con las redes neuronales potentes es que pueden «recordar» estructuras de sus conjuntos de entrenamiento en lugar de descubrir nuevas. Para comprobar si EvoSplit realmente añade información, los autores recurren a un conjunto de transportadores y receptores cuyos estados alternativos no se incluyeron en el entrenamiento original de AlphaFold2. Incluso aquí, EvoSplit recupera tanto las formas orientadas hacia el interior como las orientadas hacia el exterior, así como formas activas e inactivas distintas, con alta similitud estructural respecto a modelos experimentales. El método también escala a tareas más exploratorias: aplicado a más de cien proteínas vinculadas al cáncer humano, señala 54 candidatas que probablemente adopten múltiples conformaciones. Para algunas, como la quinasa LCK y el regulador del ciclo celular ciclina D1, EvoSplit sugiere disposiciones plausibles de dominios que evocan estructuras conocidas de proteínas relacionadas, insinuando estados no observados pero biológicamente razonables.
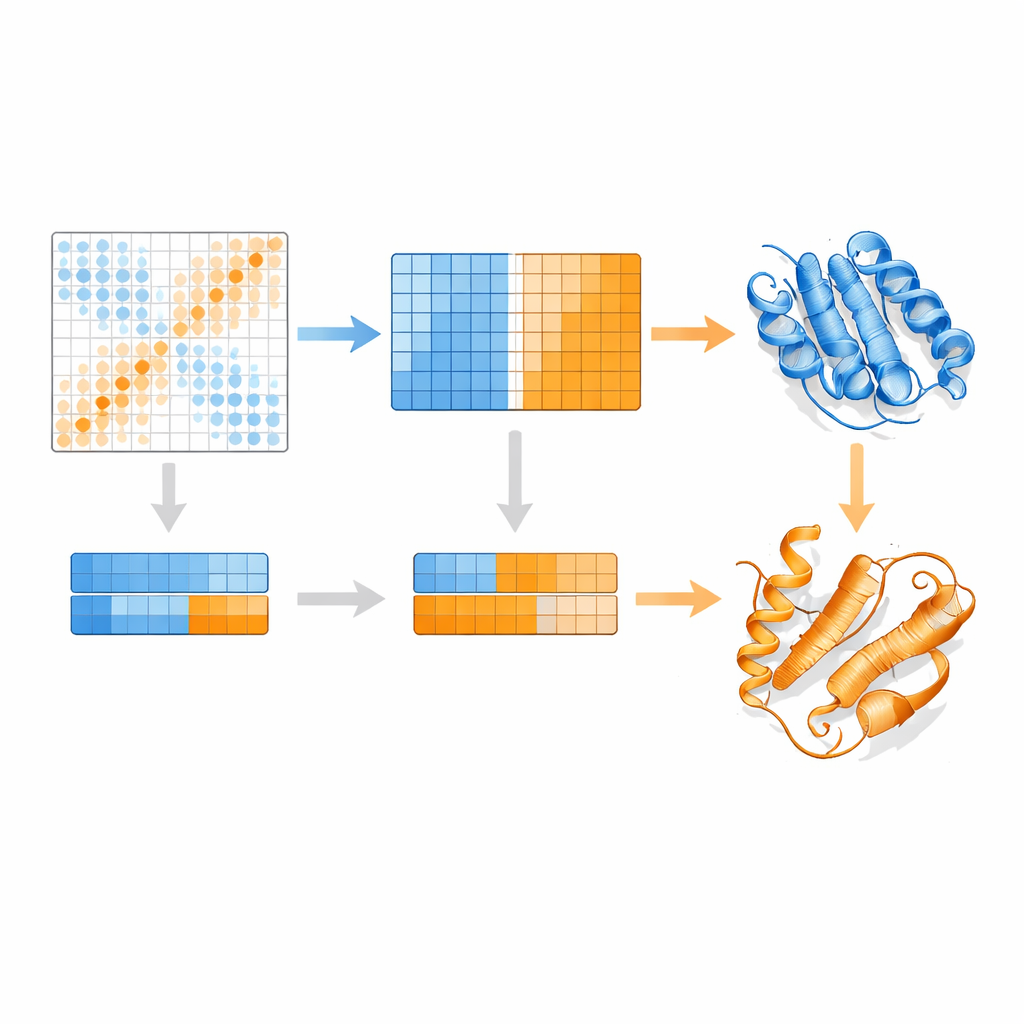
Un nuevo pliegue sorprendente en conmutadores vinculados al cáncer
Quizá el resultado más intrigante concierne a pequeñas GTPasas como HRAS y KRAS, conmutadores moleculares clásicos que se mutan con frecuencia en tumores. Estas proteínas normalmente alternan entre «encendido» y «apagado» por reorganizaciones sutiles cerca del sitio de unión del nucleótido, manteniendo el resto de su pliegue intacto. EvoSplit, sin embargo, predice repetidamente una conformación alternativa en la que una hélice cercana al inicio de la proteína se convierte en una lámina, alterando la topología general. Este patrón aparece en cinco GTPasas relacionadas, lo que sugiere que no es un accidente. Las simulaciones de este estado inusual se mantienen estables durante cientos de nanosegundos, y los análisis de acoplamientos evolutivos muestran señales de contacto distintivas que coinciden con sus contactos únicos en lámina. Cuando los autores modelan interacciones entre HRAS y varios socios conocidos, tanto la conformación clásica como la nueva forman complejos estables, pero con interfaces de contacto desplazadas, lo que implica que el pliegue alternativo podría soportar comportamientos de señalización distintos.
Qué implica esto para el diseño de fármacos futuro
Para un público no especializado, el mensaje central es que nuestras proteínas pueden albergar más formas —y por tanto más posibilidades funcionales— de las que ha revelado la predicción estructural tradicional. EvoSplit usa el reconocimiento de patrones guiado por la evolución para separar estos estados ocultos en lugar de promediarlos. Al superar a métodos anteriores en proteínas conocidas por cambiar de pliegue, descubrir estados alternativos en receptores y transportadores bien estudiados y sugerir un nuevo pliegue estable para conmutadores relacionados con el cáncer como HRAS, este trabajo sostiene que el modelado multiestado debería volverse rutinario. En términos prácticos, catálogos estructurales más ricos podrían revelar nuevos bolsillos para fármacos, explicar por qué ciertas mutaciones son dañinas y señalar vías que solo se hacen visibles cuando miramos más allá de una estructura estática única.
Cita: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Palabras clave: dinámica conformacional de proteínas, señales coevolutivas, AlphaFold2, proteínas relacionadas con el cáncer, cambio de pliegue en GTPasas