Clear Sky Science · pl
Rozplatanie koewolucyjnych ograniczeń w modelowaniu konformacyjnej heterogeniczności białek
Białka jako maszyny zmieniające kształt
Białka to drobne maszyny, które umożliwiają życie, a wiele z nich działa poprzez subtelną zmianę kształtu. Te przesunięcia mogą włączać lub wyłączać sygnały, otwierać lub zamykać molekularne bramki albo przekształcać kieszenie wiążące, na które celują leki. Tymczasem większość narzędzi komputerowych wciąż próbuje przypisać białku jedną „poprawną” strukturę, ukrywając tym samym elastyczność leżącą u podstaw zdrowia i choroby. W artykule przedstawiono EvoSplit — nową metodę odczytywania zapisu ewolucyjnego zawartego w sekwencjach białkowych, pozwalającą odkryć wiele funkcjonalnie istotnych kształtów — w tym niektóre nigdy nie zaobserwowane doświadczalnie, które mogą otworzyć nowe możliwości w odkrywaniu leków.
Dlaczego elastyczność białek ma znaczenie w medycynie
W komórkach białka rzadko pozostają nieruchome. Zginają się, skręcają, a czasem nawet przełączają fragmenty swojej struktury w odpowiedzi na zmiany temperatury, kwasowości, partnerów wiążących czy modyfikacji chemicznych. Ruchy te mogą być niewielkie, jak przesunięcie kilku łańcuchów bocznych, albo dramatyczne, gdy całe sekcje przechodzą z helisy w płaszcz. Zmiany te są kluczowe dla tego, jak receptory rozpoznają hormony, jak transportery przemieszczają cząsteczki przez błony i jak onkoproteiny napędzają rozwój nowotworu. Znając tylko jedno ujęcie białka, możemy nie zauważyć formy aktywnej, podatnej na leki kieszeni ani konformacji prowadzącej do choroby. Ustalenie dokładnego „składu ról” stabilnych kształtów białka jest więc niezbędne do zrozumienia biologii i projektowania ukierunkowanych terapii.
Odczytywanie ukrytych nut ewolucji
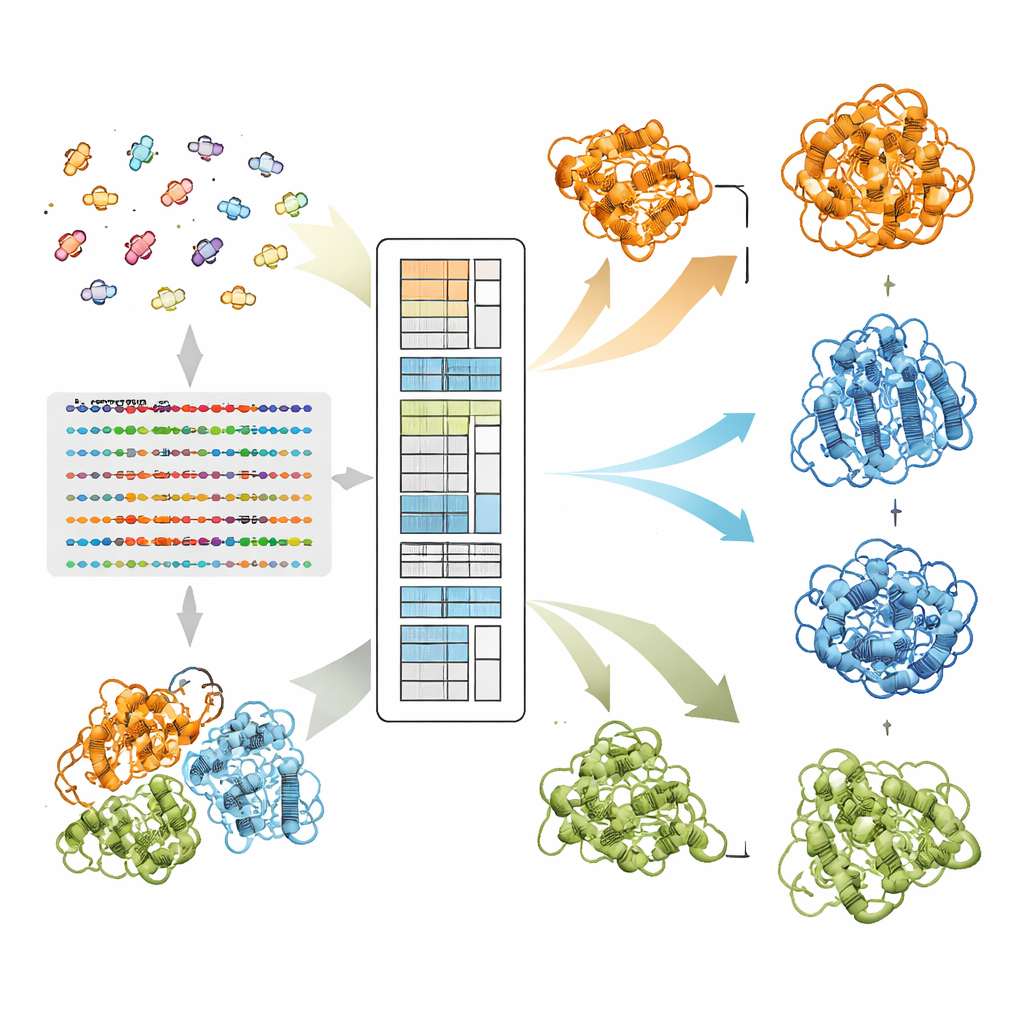
Przez miliony lat sekwencje białek ewoluowały pod presją zachowania nie tylko jednej struktury, lecz często kilku biologicznie istotnych kształtów. Gdy dwie aminokwasy zwykle mutują w skoordynowany sposób wśród spokrewnionych białek, sugeruje to, że muszą pozostawać w kontakcie przynajmniej w jednej konformacji. Nowoczesne systemy uczenia głębokiego, takie jak AlphaFold2, osiągają sukces, ponieważ wydobywają takie wzorce koewolucyjne z dużych rodzin spokrewnionych sekwencji, nazywanych wielokrotnymi wyrównaniami sekwencji. Jednak gdy białko może przyjmować więcej niż jeden fałd, sygnały dotyczące różnych stanów mieszają się, a standardowe podejścia zwykle sprowadzają je do pojedynczej, uśrednionej struktury. Wcześniejsze metody próbowały rozdzielać to przez klastrowanie sekwencji na podstawie ogólnego podobieństwa, lecz te podejścia w dużej mierze ignorowały parowe wzorce, które w rzeczywistości kodują preferencje strukturalne.
Jak EvoSplit rozdziela nakładające się kształty
Autorzy opierają się na modelu języka białkowego o nazwie MSA Transformer, który wykorzystuje mechanizm uwagi (attention), aby nauczyć się, które reszty w których sekwencjach „zwracają na siebie uwagę”. Pokazują, że dla białek z wieloma znanymi strukturami wzór uwagi każdej pojedynczej sekwencji ma tendencję do przypominania mapy kontaktów jednej konkretnej konformacji bardziej niż innej. Innymi słowy, każda sekwencja nosi odcisk swojego preferowanego kształtu. EvoSplit wykorzystuje to, stosując macierze uwagi — a nie surowe podobieństwo sekwencji — jako cechy do klastrowania wyrównania na podgrupy. Każdy klaster następnie podawany jest oddzielnie do AlphaFold2, co skutecznie daje predyktorowi struktury czystszy, specyficzny dla konformacji kontekst ewolucyjny. W badaniu obejmującym 85 białek znanych z przełączania fałdów, EvoSplit generuje modele lepiej zgodne ze strukturami eksperymentalnymi i o wyższym zaufaniu niż wiodąca metoda klastrowania oparta na sekwencjach, zwłaszcza dla rzadziej próbkowanego stanu.
Odkrywanie nowych stanów poza danymi treningowymi
Kluczowym zmartwieniem przy potężnych sieciach neuronowych jest to, że mogą one po prostu „zapamiętywać” struktury z zestawów treningowych, zamiast odkrywać nowe. Aby sprawdzić, czy EvoSplit naprawdę wnosi nową informację, autorzy sięgają do zbioru transporterów i receptorów, których alternatywne stany nie były uwzględnione w oryginalnym treningu AlphaFold2. Nawet w tym przypadku EvoSplit odtwarza zarówno formy zwrócone do wnętrza, jak i na zewnątrz, a także wyraźne konformacje aktywne i nieaktywne, o wysokim podobieństwie strukturalnym do modeli eksperymentalnych. Metoda skaluje się także do bardziej eksploracyjnych zadań: zastosowana do ponad stu białek powiązanych z nowotworami ludzkimi, wskazuje 54 kandydatów, które prawdopodobnie przyjmują wiele konformacji. Dla niektórych z nich, takich jak kinaza LCK i regulator cyklu komórkowego cyklina D1, EvoSplit sugeruje wiarygodne ułożenia domen korespondujące ze znanymi strukturami spokrewnionych białek, co sugeruje niezaobserwowane, ale biochemicznie prawdopodobne stany.
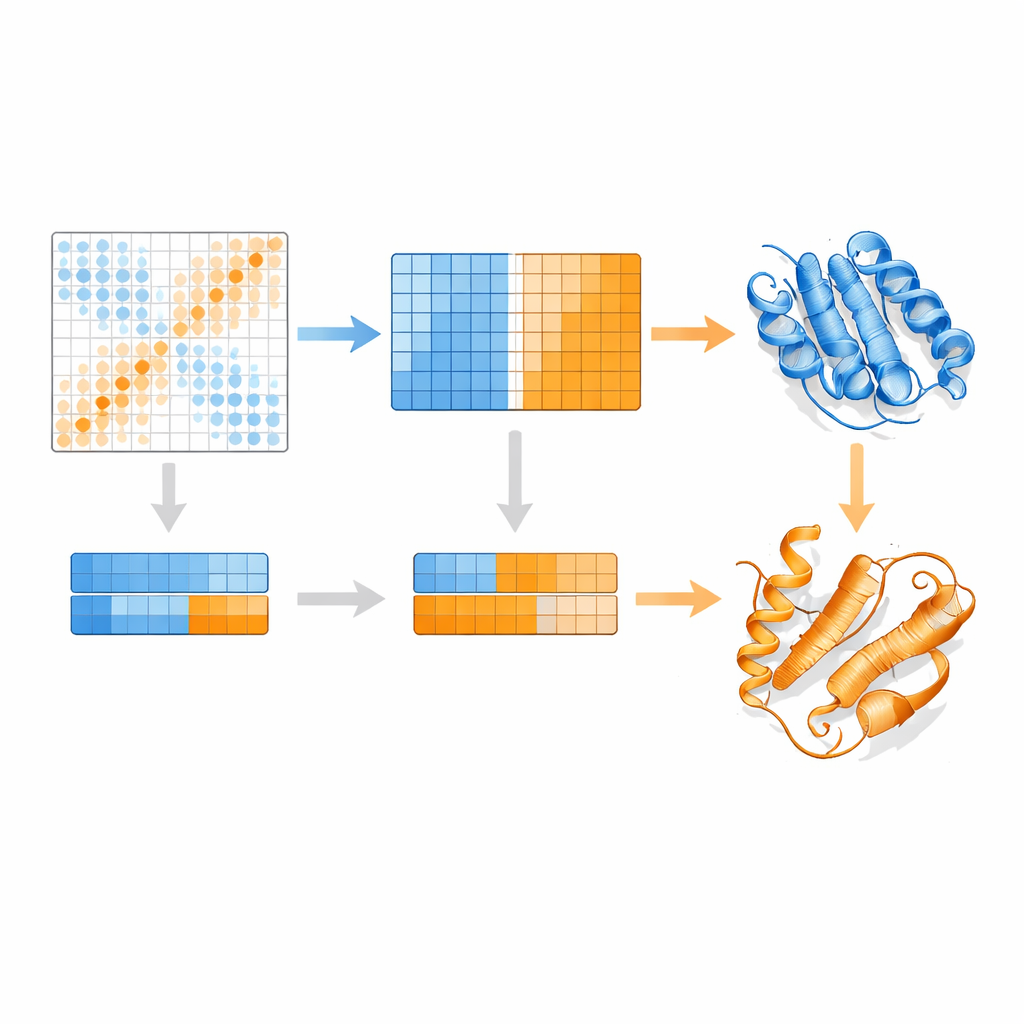
Zaskakujący nowy fałd w przełącznikach powiązanych z rakiem
Być może najbardziej intrygujący rezultat dotyczy małych GTPaz, takich jak HRAS i KRAS, klasycznych molekularnych przełączników często mutowanych w nowotworach. Te białka zwykle przełączają się między „włączonym” a „wyłączonym” stanem przez subtelne przearanżowania w pobliżu miejsca wiążącego nukleotyd, zachowując jednocześnie resztę fałdu. EvoSplit jednak wielokrotnie przewiduje alternatywną konformację, w której jedna helisa blisko początku białka przekształca się w płaszcz, zmieniając ogólną topologię. Wzorzec ten pojawia się w pięciu spokrewnionych GTPazach, co sugeruje, że nie jest to przypadek. Symulacje tej nietypowej formy pozostają stabilne przez setki nanosekund, a analizy sprzężeń ewolucyjnych wykazują odrębne sygnały kontaktów pasujące do unikalnych kontaktów płaszcza. Gdy autorzy modelują interakcje między HRAS a kilkoma znanymi partnerami, zarówno klasyczna, jak i nowa konformacja tworzą stabilne kompleksy, ale z przesuniętymi interfejsami kontaktów, co sugeruje, że alternatywny fałd mógłby wspierać różne zachowania sygnalizacyjne.
Co to oznacza dla przyszłego projektowania leków
Dla osoby nie będącej specjalistą główne przesłanie jest takie, że nasze białka mogą skrywać więcej kształtów — a zatem więcej funkcjonalnych możliwości — niż ujawnia tradycyjne przewidywanie struktur. EvoSplit wykorzystuje ewolucyjnie sterowane rozpoznawanie wzorców, aby oddzielić te ukryte stany zamiast je uśredniać. Przewyższając wcześniejsze metody na znanych białkach przełączających fałdy, odkrywając alternatywne stany w dobrze zbadanych receptorach i transporterach oraz sugerując nowy, stabilny fałd dla przełączników związanych z rakiem, takich jak HRAS, praca ta argumentuje, że modelowanie wielostanowe powinno stać się rutyną. W praktyce bogatsze katalogi struktur mogą uwydatnić nowe kieszenie dla leków, wyjaśnić, dlaczego niektóre mutacje są szkodliwe, i wskazać ścieżki ujawniające się dopiero wtedy, gdy spojrzymy poza pojedynczą, statyczną strukturę.
Cytowanie: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Słowa kluczowe: dynamiczna konformacja białek, sygnały koewolucyjne, AlphaFold2, białka związane z rakiem, przełączanie fałdu GTPazy