Clear Sky Science · de
Entflechtung koevolutionärer Zwänge zur Modellierung konformationeller Heterogenität von Proteinen
Proteine als Gestaltwandelnde Maschinen
Proteine sind die winzigen Maschinen, die das Leben ermöglichen, und viele von ihnen funktionieren, indem sie ihre Form subtil verändern. Diese Veränderungen können Signale an- oder ausschalten, molekulare Tore öffnen oder schließen oder Bindungstaschen umgestalten, die von Arzneistoffen anvisiert werden. Dennoch versuchen die meisten Computerwerkzeuge immer noch, jedem Protein nur eine „richtige“ Struktur zuzuweisen und verbergen damit genau jene Flexibilität, die Gesundheit und Krankheit zugrunde liegt. Dieses Papier stellt EvoSplit vor, eine neue Methode, um die in Proteinsequenzen gespeicherte evolutionäre Historie zu lesen und mehrere funktionell relevante Formen aufzudecken — einschließlich solcher, die in Experimenten noch nie beobachtet wurden und neue Wege für die Wirkstoffforschung eröffnen könnten.
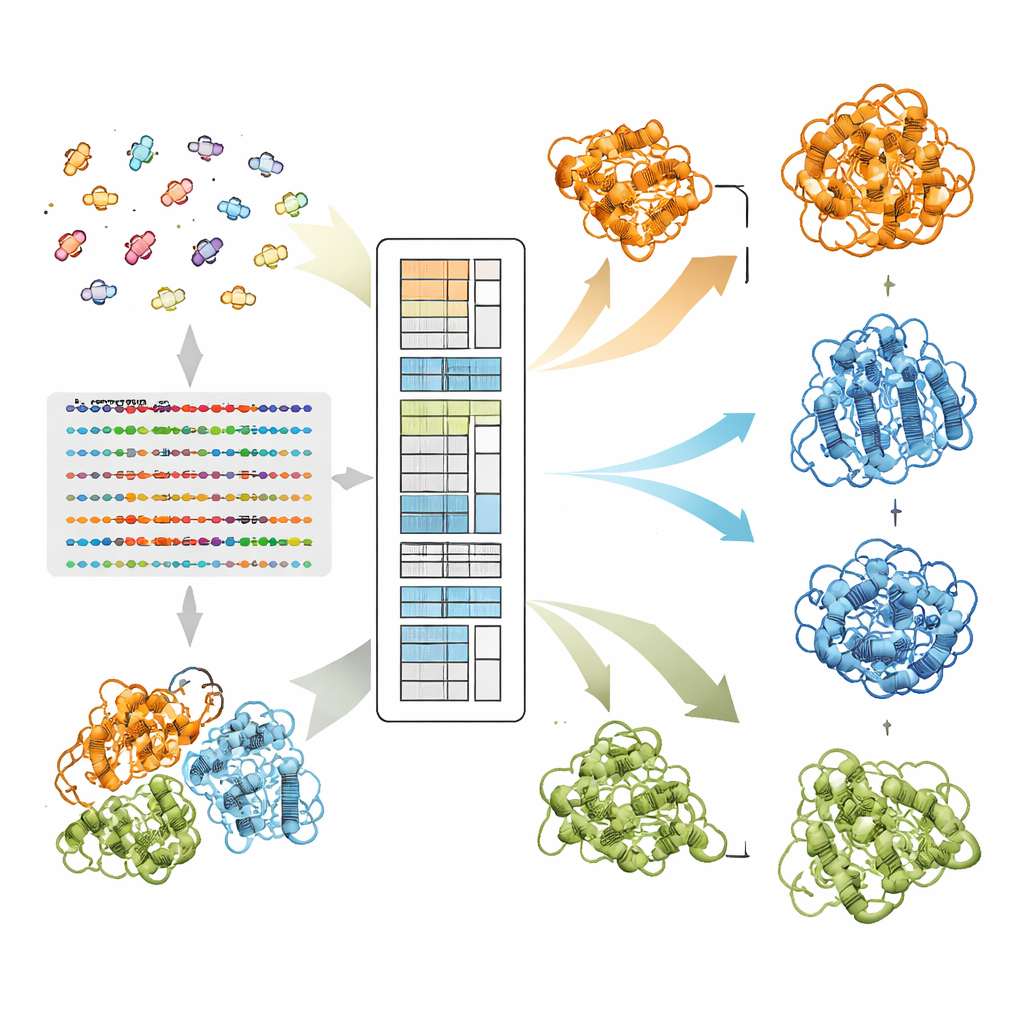
Warum Proteinfexibilität für die Medizin wichtig ist
In unseren Zellen bleiben Proteine selten starr. Sie biegen sich, verdrehen sich und falten manchmal Teile von sich selbst als Reaktion auf Änderungen der Temperatur, des pH-Werts, von Bindungspartnern oder chemischen Modifikationen um. Solche Bewegungen können klein sein, wie das Verschieben einiger Seitengruppen, oder dramatisch, wenn ganze Abschnitte von einer Helix zu einem Blatt umschlagen. Diese Änderungen sind zentral dafür, wie Rezeptoren Hormone wahrnehmen, wie Transporter Moleküle über Membranen bewegen und wie Onkoproteine Krebs antreiben. Kennt man nur einen Schnappschuss eines Proteins, kann man die aktive Form, die druggbare Tasche oder die Konformation, die zur Krankheit führt, übersehen. Eine genaue „Besetzungsliste“ der stabilen Formen eines Proteins zu erfassen, ist daher entscheidend, um Biologie zu verstehen und gezielte Therapien zu entwickeln.
Die verborgenen Notizen der Evolution lesen
Über Millionen von Jahren haben sich Proteinsequenzen unter dem Druck entwickelt, nicht nur eine einzelne Struktur, sondern häufig mehrere biologisch relevante Formen zu erhalten. Wenn zwei Aminosäuren dazu neigen, in koordinierter Weise über verwandte Proteine hinweg zu mutieren, deutet das darauf hin, dass sie in mindestens einer Konformation in Kontakt bleiben müssen. Moderne Deep-Learning-Systeme wie AlphaFold2 sind erfolgreich, weil sie solche koevolutionären Muster aus großen Familien verwandter Sequenzen — sogenannten Multiple Sequence Alignments — auswerten. Wenn ein Protein jedoch mehr als eine Faltung annehmen kann, vermischen sich die Signale für die unterschiedlichen Zustände, und Standardansätze reduzieren sie meist auf eine einzige, gemittelte Struktur. Frühere Methoden versuchten, dies durch das Clustern von Sequenzen nach ihrer allgemeinen Ähnlichkeit zu trennen, übersahen dabei jedoch weitgehend die paarweisen Muster, die tatsächlich strukturelle Präferenzen codieren.
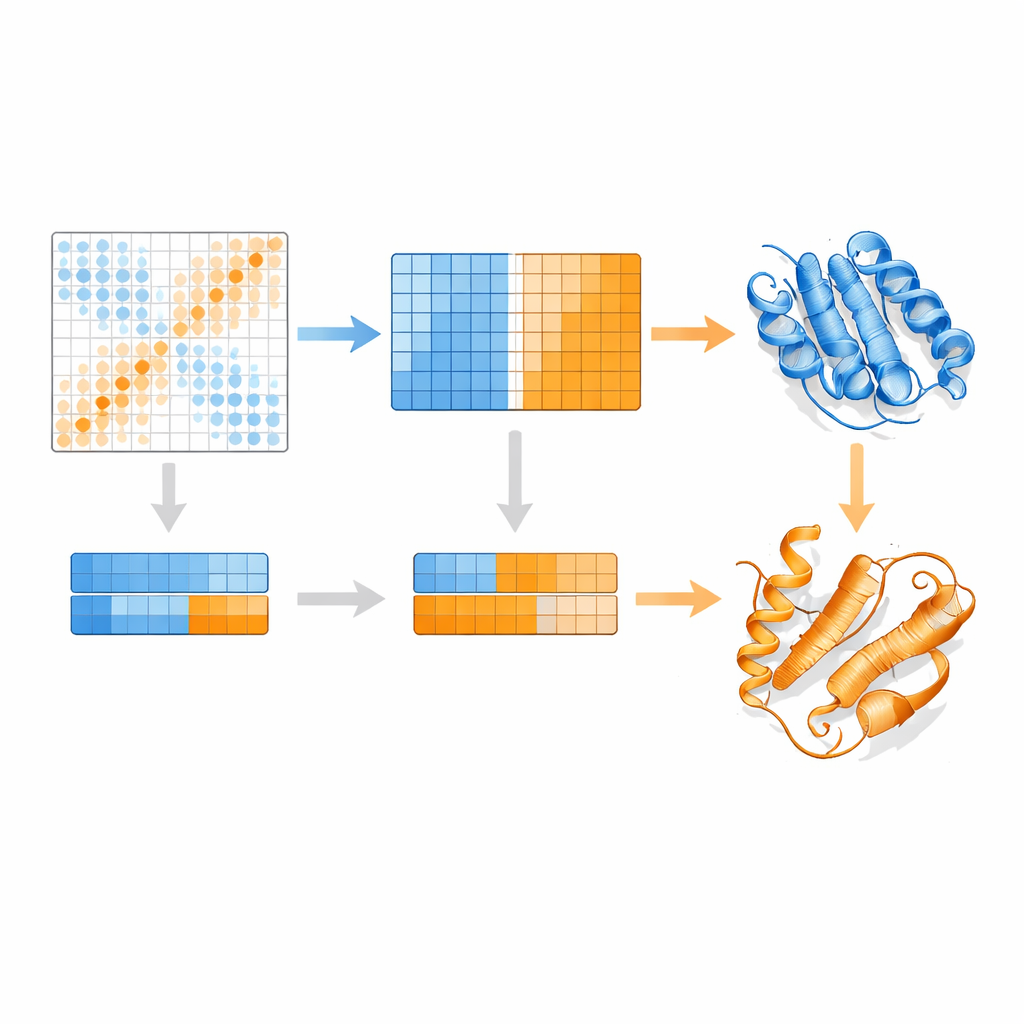
Wie EvoSplit überlappende Formen auseinanderzieht
Die Autoren bauen auf einem Protein-Sprachmodell namens MSA Transformer auf, das einen Attention-Mechanismus verwendet, um zu lernen, welche Reste in welchen Sequenzen „aufeinander achten“. Sie zeigen, dass bei Proteinen mit mehreren bekannten Strukturen das Aufmerksamkeitsmuster jeder einzelnen Sequenz eher der Kontaktkarte einer spezifischen Konformation gleicht als der anderen. Mit anderen Worten trägt jede Sequenz einen Fingerabdruck ihrer bevorzugten Form. EvoSplit nutzt dies, indem es die Attention-Matrizen — nicht die rohe Sequenzähnlichkeit — als Merkmale zum Clustern des Alignments in Untergruppen verwendet. Jeder Cluster wird dann separat in AlphaFold2 eingespeist, wodurch der Strukturvorhersage gewissermaßen ein saubererer, konformationsspezifischer evolutionärer Hinweis gegeben wird. Bei 85 Proteinen, die für Faltungsumschläge bekannt sind, liefert EvoSplit Modelle, die besser mit experimentellen Strukturen übereinstimmen und mit höherem Vertrauen bewertet werden als eine führende sequenzbasierte Clustering-Methode, besonders für den seltener beobachteten Zustand.
Neue Zustände jenseits der Trainingsdaten finden
Eine zentrale Sorge bei leistungsfähigen neuronalen Netzen ist, dass sie möglicherweise einfach Strukturen aus ihren Trainingsdaten „auswendig gelernt“ haben, statt neue zu entdecken. Um zu prüfen, ob EvoSplit tatsächlich zusätzliche Information liefert, wenden die Autoren die Methode auf eine Reihe von Transportern und Rezeptoren an, deren alternative Zustände nicht in AlphaFold2s ursprünglichem Training enthalten waren. Selbst hier rekonstruiert EvoSplit sowohl eingangs- als auch auswärtsgerichtete Formen sowie unterschiedliche aktive und inaktive Strukturen mit hoher struktureller Ähnlichkeit zu experimentellen Modellen. Die Methode skaliert auch zu explorativeren Aufgaben: Auf über hundert mit menschlichen Krebserkrankungen verknüpfte Proteine angewandt, identifiziert sie 54 Kandidaten, die wahrscheinlich mehrere Konformationen annehmen. Für einige, wie die Kinase LCK und den Zellzyklusregulator Cyclin D1, schlägt EvoSplit plausible Anordnungen von Domänen vor, die bekannten Strukturen verwandter Proteine ähneln und auf bislang unobservierte, aber biophysikalisch sinnvolle Zustände hinweisen.
Eine überraschende neue Faltung in krebsverknüpften Schaltern
Vielleicht ist das faszinierendste Ergebnis die Beobachtung bei kleinen GTPasen wie HRAS und KRAS, klassischen molekularen Schaltern, die häufig in Tumoren mutiert sind. Diese Proteine schalten normalerweise zwischen „an“ und „aus“ durch subtile Umordnungen in der Nähe der Nukleotidbindungsstelle, während der Rest ihrer Faltung intakt bleibt. EvoSplit sagt jedoch wiederholt eine alternative Konformation voraus, in der eine Helix nahe dem N-Terminus des Proteins in ein Blatt übergeht und dadurch die Gesamt-Topologie verändert. Dieses Muster tritt bei fünf verwandten GTPasen auf, was darauf hindeutet, dass es kein Zufall ist. Simulationen dieses ungewöhnlichen Zustands bleiben über Hunderte von Nanosekunden stabil, und Analysen evolutionärer Kopplungen zeigen eindeutige Kontakt-Signale, die mit den einzigartigen Blattkontakten übereinstimmen. Wenn die Autoren Wechselwirkungen zwischen HRAS und mehreren bekannten Partnern modellieren, bilden sowohl die klassische als auch die neue Konformation stabile Komplexe, jedoch mit verschobenen Kontaktoberflächen, was darauf hindeutet, dass die alternative Faltung unterschiedliche Signalverhaltensweisen unterstützen könnte.
Was das für zukünftiges Wirkstoffdesign bedeutet
Für Nicht-Spezialisten lautet die Kernbotschaft, dass unsere Proteine mehr Formen — und damit mehr funktionelle Möglichkeiten — beherbergen können, als traditionelle Strukturvorhersagen gezeigt haben. EvoSplit nutzt evolutionär geleitete Mustererkennung, um diese verborgenen Zustände zu trennen, statt sie zu verwischen. Indem es frühere Methoden bei bekannten Faltungsumschlägen übertrifft, alternative Zustände in gut untersuchten Rezeptoren und Transportern entdeckt und eine neue, stabile Faltung für krebsrelevante Schalter wie HRAS vorschlägt, plädiert diese Arbeit dafür, dass Mehrzustands-Modellierung zur Routine werden sollte. Praktisch könnten umfangreichere Strukturkataloge neue Taschen für Arzneistoffe hervorheben, erklären, warum bestimmte Mutationen schädlich sind, und auf Signalwege hinweisen, die erst sichtbar werden, wenn man über eine einzelne statische Struktur hinausblickt.
Zitation: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Schlüsselwörter: konformationelle Dynamik von Proteinen, koevolutionäre Signale, AlphaFold2, krebsbezogene Proteine, GTPase-Faltungsumschlag