Clear Sky Science · en
Disentangling coevolutionary constraints for modeling protein conformational heterogeneity
Proteins as Shape-Shifting Machines
Proteins are the tiny machines that make life possible, and many of them work by subtly changing shape. These shifts can turn signals on or off, open or close molecular gates, or reshape binding pockets that drugs aim to hit. Yet most computer tools still try to assign each protein just one "correct" structure, hiding the very flexibility that underlies health and disease. This paper introduces EvoSplit, a new way to read the evolutionary record encoded in protein sequences to uncover multiple, functionally important shapes—including some that have never been seen in experiments and may open fresh avenues for drug discovery.
Why Protein Flexibility Matters for Medicine
Inside our cells, proteins rarely sit still. They bend, twist, and sometimes even refold parts of themselves in response to changes in temperature, acidity, binding partners, or chemical modifications. Such motions can be small, like a few side chains shifting, or dramatic, like entire sections flipping from a helix to a sheet. These changes are central to how receptors sense hormones, how transporters move molecules across membranes, and how oncoproteins drive cancer. If we only know one snapshot of a protein, we may miss the active form, the druggable pocket, or the conformation that leads to disease. Capturing an accurate "cast list" of a protein’s stable shapes is therefore crucial for understanding biology and designing targeted therapies.
Reading Evolution’s Hidden Notes
Over millions of years, protein sequences have evolved under pressure to preserve not just a single structure, but often several biologically relevant shapes. When two amino acids tend to mutate in a coordinated way across related proteins, it hints that they must stay in contact in at least one conformation. Modern deep-learning systems such as AlphaFold2 excel because they mine such coevolutionary patterns from large families of related sequences, known as multiple sequence alignments. However, when a protein can adopt more than one fold, the signals for different states become mixed together, and standard approaches usually collapse them into a single, averaged structure. Earlier methods tried to tease this apart by clustering sequences based on their overall similarity, but those approaches largely ignored the pairwise patterns that actually encode structural preferences.
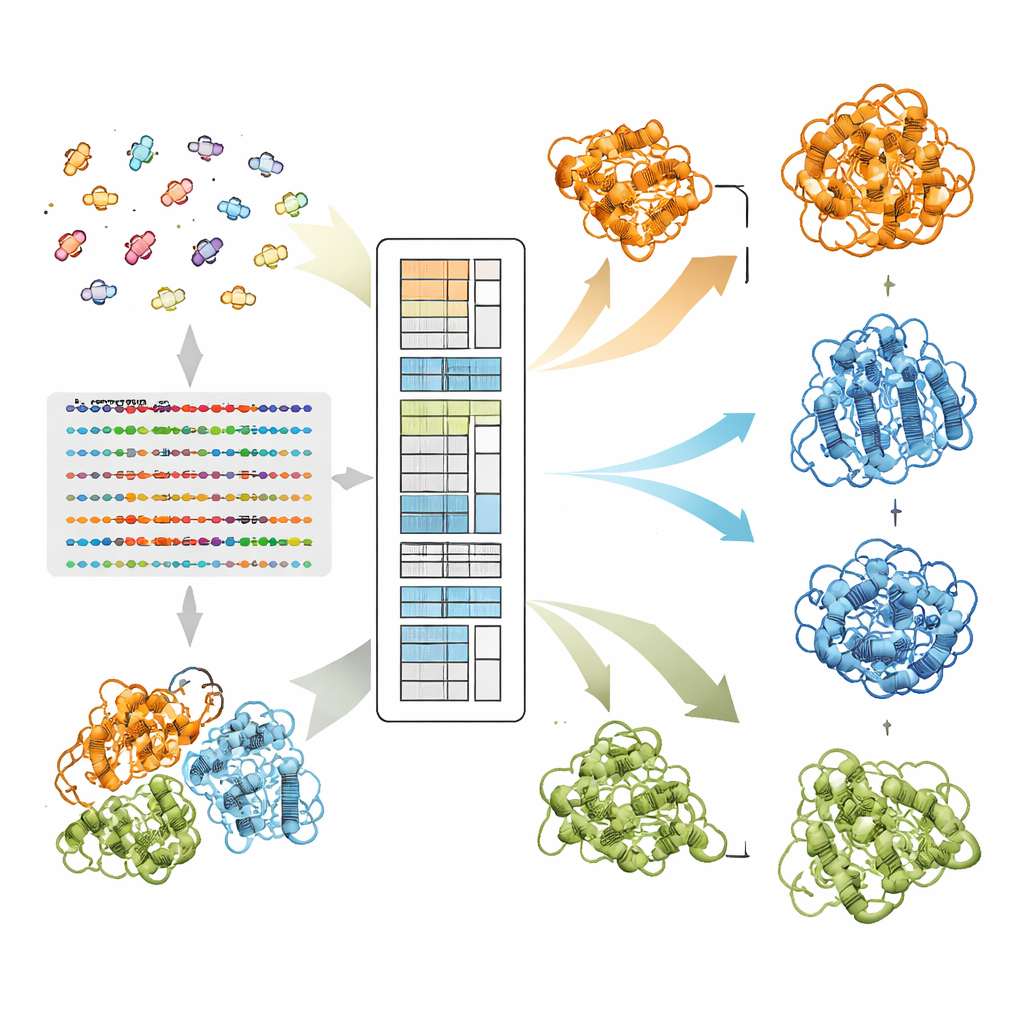
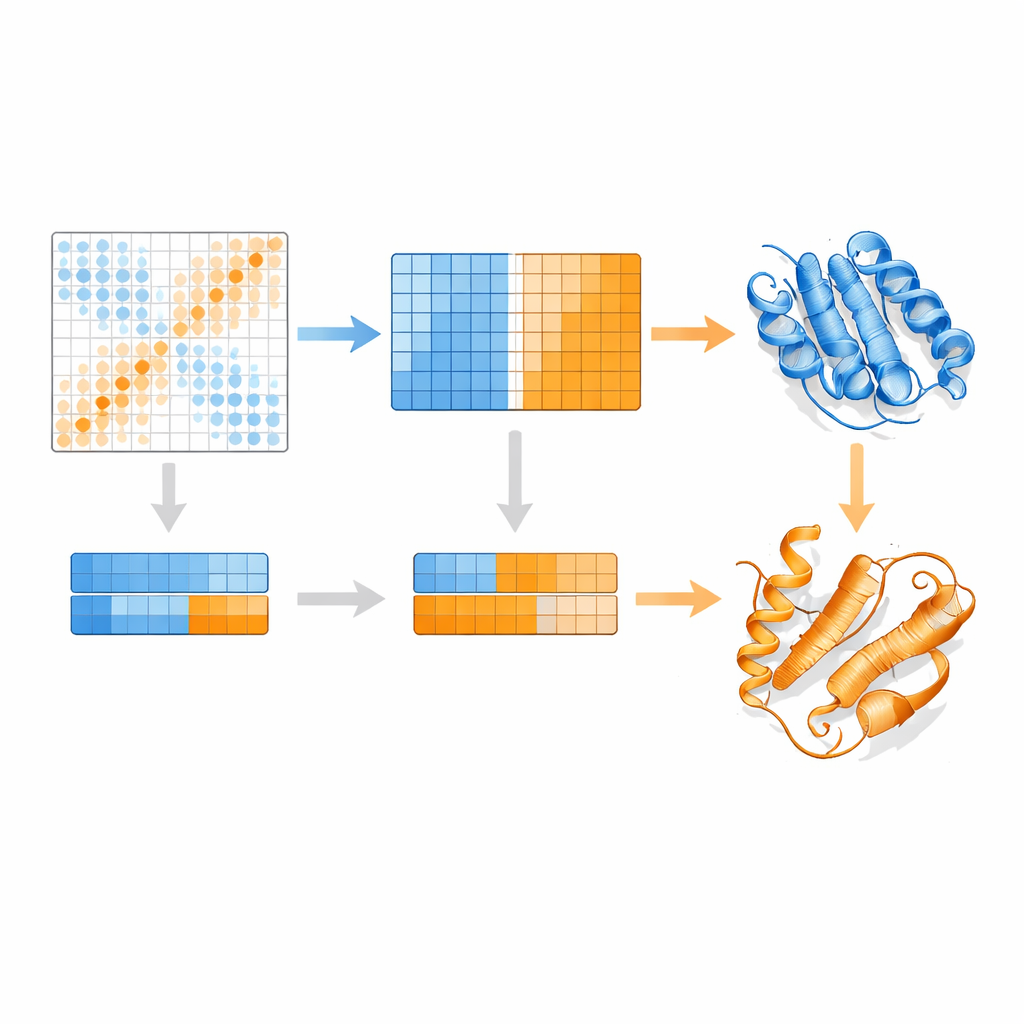
How EvoSplit Pulls Apart Overlapping Shapes
The authors build on a protein language model called MSA Transformer, which uses an attention mechanism to learn which residues in which sequences "pay attention" to one another. They show that, for proteins with multiple known structures, the attention pattern of each individual sequence tends to resemble the contact map of one specific conformation more than the other. In other words, each sequence carries a fingerprint of its favorite shape. EvoSplit harnesses this by using the attention matrices—not raw sequence similarity—as features for clustering the alignment into subgroups. Each cluster is then fed separately into AlphaFold2, effectively giving the structure predictor a cleaner, conformation-specific evolutionary prompt. Across 85 proteins known to switch folds, EvoSplit produces models that agree better with experimental structures and with higher confidence than a leading sequence-based clustering method, especially for the more rarely sampled state.
Finding New States Beyond the Training Data
A key concern with powerful neural networks is that they may simply "remember" structures from their training sets rather than discover new ones. To test whether EvoSplit truly adds information, the authors turn to a set of transporters and receptors whose alternative states were not included in AlphaFold2’s original training. Even here, EvoSplit recovers both inward- and outward-facing forms, as well as distinct active and inactive shapes, with high structural similarity to experimental models. The method also scales to more exploratory tasks: applied to over a hundred proteins linked to human cancers, it flags 54 candidates likely to adopt multiple conformations. For some, such as the kinase LCK and the cell-cycle regulator cyclin D1, EvoSplit suggests plausible arrangements of domains that echo known structures from related proteins, hinting at unobserved but biophysically reasonable states.
A Surprising New Fold in Cancer-Linked Switches
Perhaps the most intriguing result concerns small GTPases such as HRAS and KRAS, classic molecular switches frequently mutated in tumors. These proteins normally toggle between "on" and "off" by subtle rearrangements near the nucleotide-binding site while keeping the rest of their fold intact. EvoSplit, however, repeatedly predicts an alternative conformation in which one helix near the protein’s beginning converts into a sheet, altering the overall topology. This pattern appears across five related GTPases, suggesting it is not a fluke. Simulations of this unusual state remain stable over hundreds of nanoseconds, and analyses of evolutionary couplings show distinct contact signals that line up with its unique sheet contacts. When the authors model interactions between HRAS and several known partners, both the classic and the new conformation form stable complexes, but with shifted contact interfaces, implying that the alternative fold could support different signaling behaviors.
What This Means for Future Drug Design
To a non-specialist, the core message is that our proteins may harbor more shapes—and therefore more functional possibilities—than traditional structure prediction has revealed. EvoSplit uses evolution-guided pattern recognition to separate these hidden states instead of averaging them away. By outperforming earlier methods on known fold-switching proteins, discovering alternative states in well-studied receptors and transporters, and suggesting a new, stable fold for cancer-related switches like HRAS, this work argues that multi-state modeling should become routine. In practical terms, richer structural catalogs could highlight new pockets for drugs, explain why certain mutations are harmful, and point to pathways that only become visible when we look beyond a single static structure.
Citation: Li, S., Zhang, C., Kong, L. et al. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity. Commun Chem 9, 146 (2026). https://doi.org/10.1038/s42004-026-01940-9
Keywords: protein conformational dynamics, coevolutionary signals, AlphaFold2, cancer-related proteins, GTPase fold switching