Clear Sky Science · ru
Сквозной многозадачный конвейер на основе глубокого внимания для предсказания свойств пептидов с оценкой неопределённости по данным масс‑спектрометрии
Почему это исследование важно для здоровья и биологии
Современные биомедицинские исследования в большой степени опираются на масс‑спектрометрию, чтобы определить, какие белки присутствуют в клетках и тканях. Тем не менее, несмотря на мощные приборы и большие базы данных, удивительная часть данных остаётся неинтерпретированной, особенно для редких или необычных белков, которые могут играть ключевую роль в таких заболеваниях, как рак или неврологические расстройства. В этой статье представлен ProteoRift — система машинного обучения, которая помогает раскрыть больше скрытой информации, предсказывая ключевые свойства фрагментов белков напрямую по сырым данным и одновременно сообщая учёным, насколько уверенны эти предсказания.

Узкое место в чтении «отпечатков» белков
Масс‑спектрометрия работает путём разрыва белков на более мелкие части, называемые пептидами, и измерения массы получившихся фрагментов. Стандартное программное обеспечение затем ищет последовательности пептидов в больших белковых базах данных, калкулируя массу и сверяя её с наблюдаемым спектром. Чтобы сделать такой поиск вычислительно осуществимым, большинство инструментов применяют простое правило: рассматриваются только кандидаты, чья суммарная масса близка к измеренному значению. Такое фильтрование по массе ускоряет работу, но имеет и обратную сторону. Если масса слегка ошибочно определена или если пептид несёт неожиданную химическую модификацию, правильный ответ может быть исключён ещё до рассмотрения, что увеличивает долю нераспознанных спектров и создаёт смещение в сторону более распространённых и «хорошо себя ведущих» пептидов.
Более умный способ сужать поиск
ProteoRift предлагает иную стратегию: вместо фильтрации кандидатов только по массе он учится извлекать более богатую информацию из каждого спектра ещё до поиска по базе. Система построена вокруг глубокой нейронной сети с механизмом внимания, которая принимает на вход шаблон пиков в спектре вместе с базовыми параметрами съёма. На основании этого она одновременно предсказывает три свойства исходного пептида: его длину, число несмытных разрезов (missed cleavages) при подготовке образца и факт наличия модификаций. Поскольку эти задачи взаимосвязаны, совместное обучение способствует формированию устойчивого внутреннего представления спектров и повышает способность модели к обобщению на новые данные.
Преобразование предсказаний в более быстрые и экономные поиски
Чтобы использовать эти предсказания на практике, авторы интегрируют ProteoRift в сквозной конвейер вместе с ранее разработанным инструментом SpeCollate, который сопоставляет спектры и последовательности пептидов в эмбеддинговом пространстве. Сначала ProteoRift относит каждый спектр к классу, определённому диапазоном длины, числом несмытных разрезов и статусом модификации. Пептиды в базе данных аналогично группируются по известным свойствам. Поисковый движок затем сравнивает спектры только с пептидами из того же класса, вместо того чтобы просматривать все пептиды с похожей массой. На различных человеческих и микробиомных наборах данных такое целевое фильтрование теоретически сокращает пространство кандидатов более чем на 90% и даёт практический выигрыш в скорости примерно в 8–12 раз по сравнению с фильтрами, основанными только на массе, при сопоставимом числе уверенно идентифицированных пептидов. В некоторых очень больших протеогеномных и мета‑протеомных базах скорость может быть ещё выше — в отдельных тестах превышая 40‑кратное ускорение.
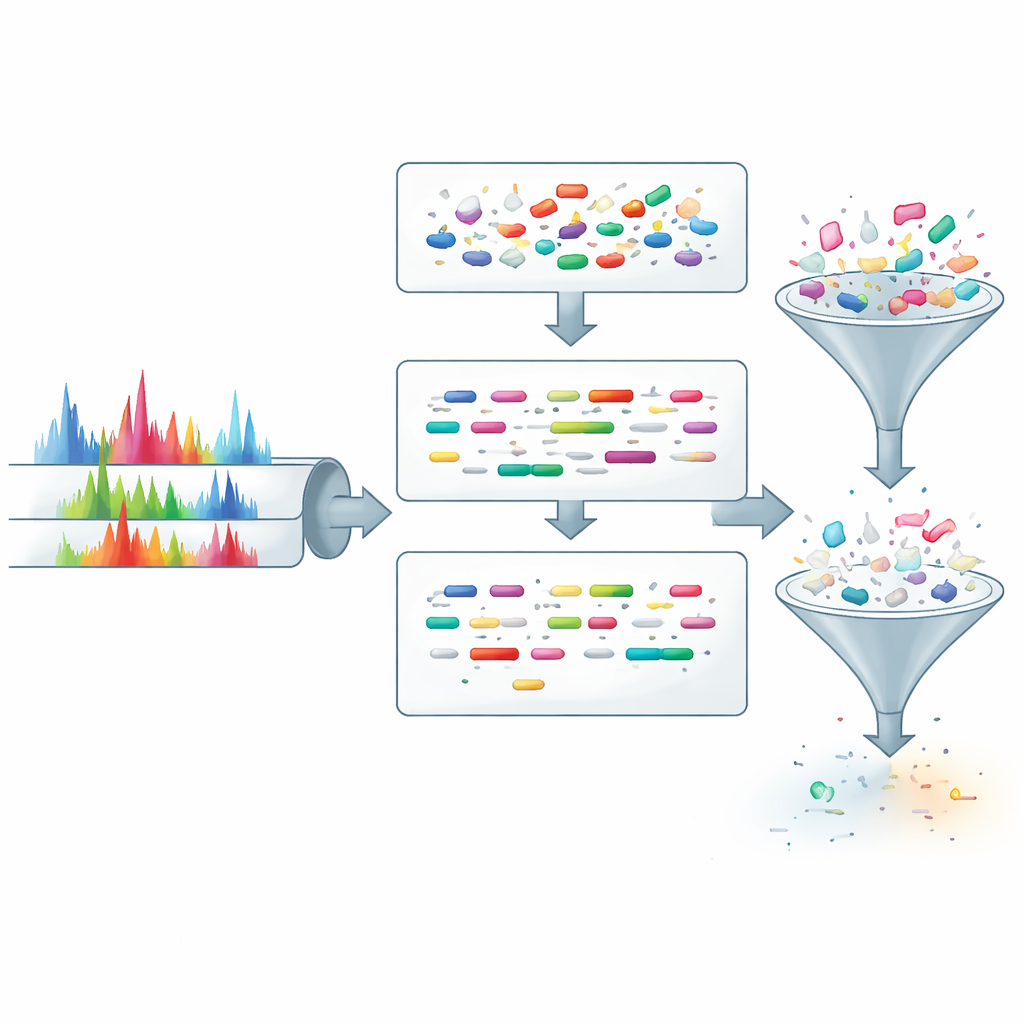
Понимание, когда модель может ошибаться
Поскольку системы машинного обучения часто воспринимаются как «чёрные ящики», авторы также разрабатывают метрики неопределённости, адаптированные под данные масс‑спектрометрии. Они исследуют, насколько меняется внутреннее представление спектра при контролируемых искажениях, насколько плотно оно окружено похожими примерами из обучающей выборки и насколько хорошо структура исходных данных сохраняется в выученном пространстве. Эти три показателя отражают разные аспекты неопределённости: шум в самих измерениях и пробелы в том, что модель видела во время обучения. В комбинации они позволяют с высокой точностью отличать знакомые данные от незнакомых и помогают отмечать случаи, когда совпадение пептида с наивысшим счётом, скорее всего, корректно.
Что это означает для будущих открытий
Проще говоря, ProteoRift функционирует как умный «шлюз», который смотрит на спектр и говорит: «это, вероятно, короткий немодифицированный пептид с одним разрезом» или «похоже на более длинный и модифицированный», и пропускает в детальный поиск только подходящих кандидатов. Это значительно ускоряет анализ без существенной потери точности, даже на сложных или очень больших белковых базах. Одновременно метрики неопределённости дают исследователям более ясное представление о том, когда результат можно доверять, а когда требуется больше данных или донастройка модели. В совокупности эти достижения могут помочь вывести масс‑спектрометрию за пределы нынешнего фокуса на массовых, хорошо изученных белках и открыть новые возможности для изучения редких и модифицированных пептидов, которые часто содержат наиболее интересные биологические подсказки.
Цитирование: Tariq, U., Shabbir, B. & Saeed, F. End-to-end deep attention-based multitask pipeline for predicting uncertainty-quantified peptide properties from mass spectrometry data. Sci Rep 16, 13331 (2026). https://doi.org/10.1038/s41598-026-43215-2
Ключевые слова: протеомика, масс‑спектрометрия, глубокое обучение, идентификация пептидов, оценка неопределённости